Unique Gut Microbiome Signatures Depict Diet-Versus Genetically Induced Obesity in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
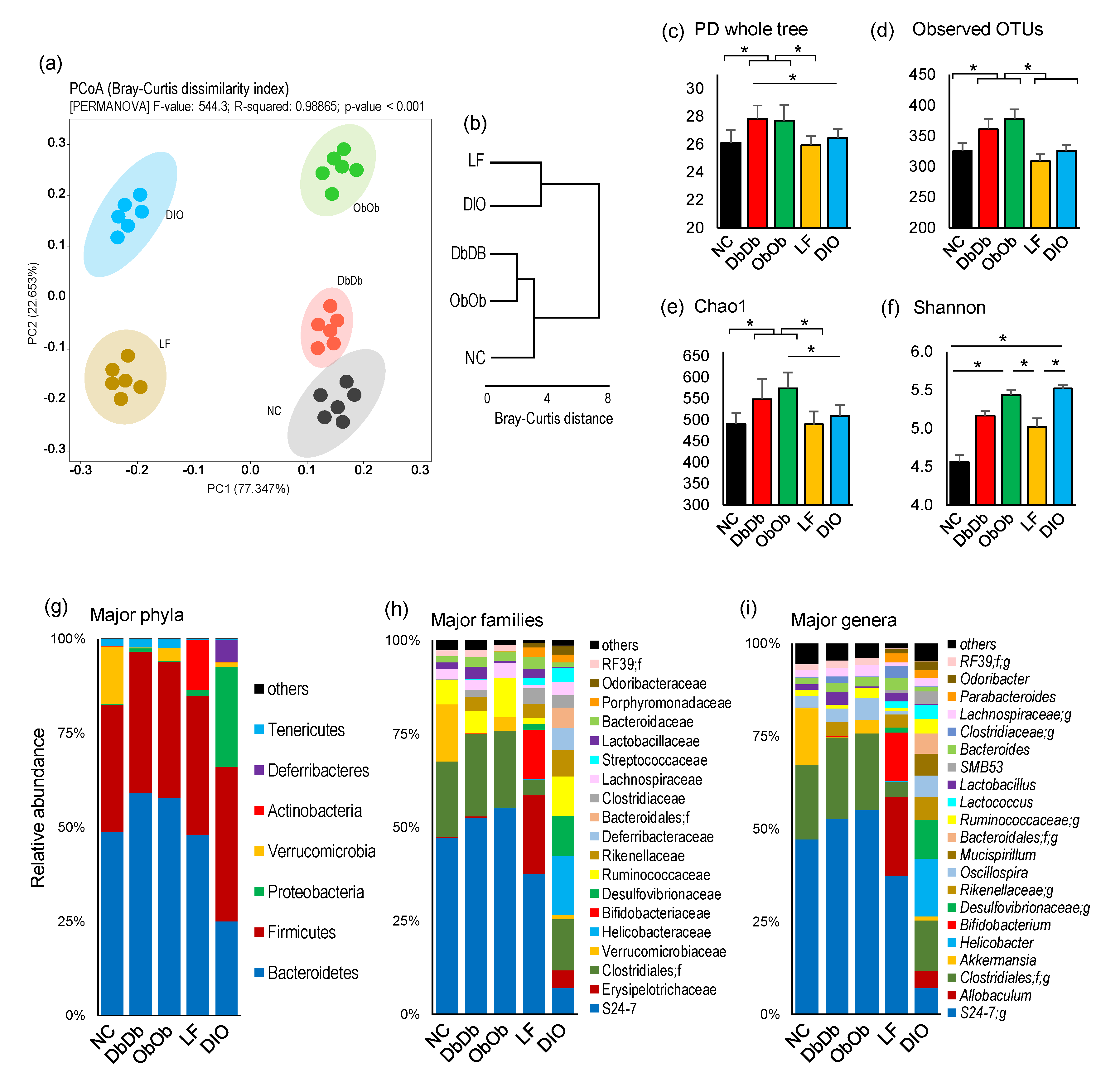
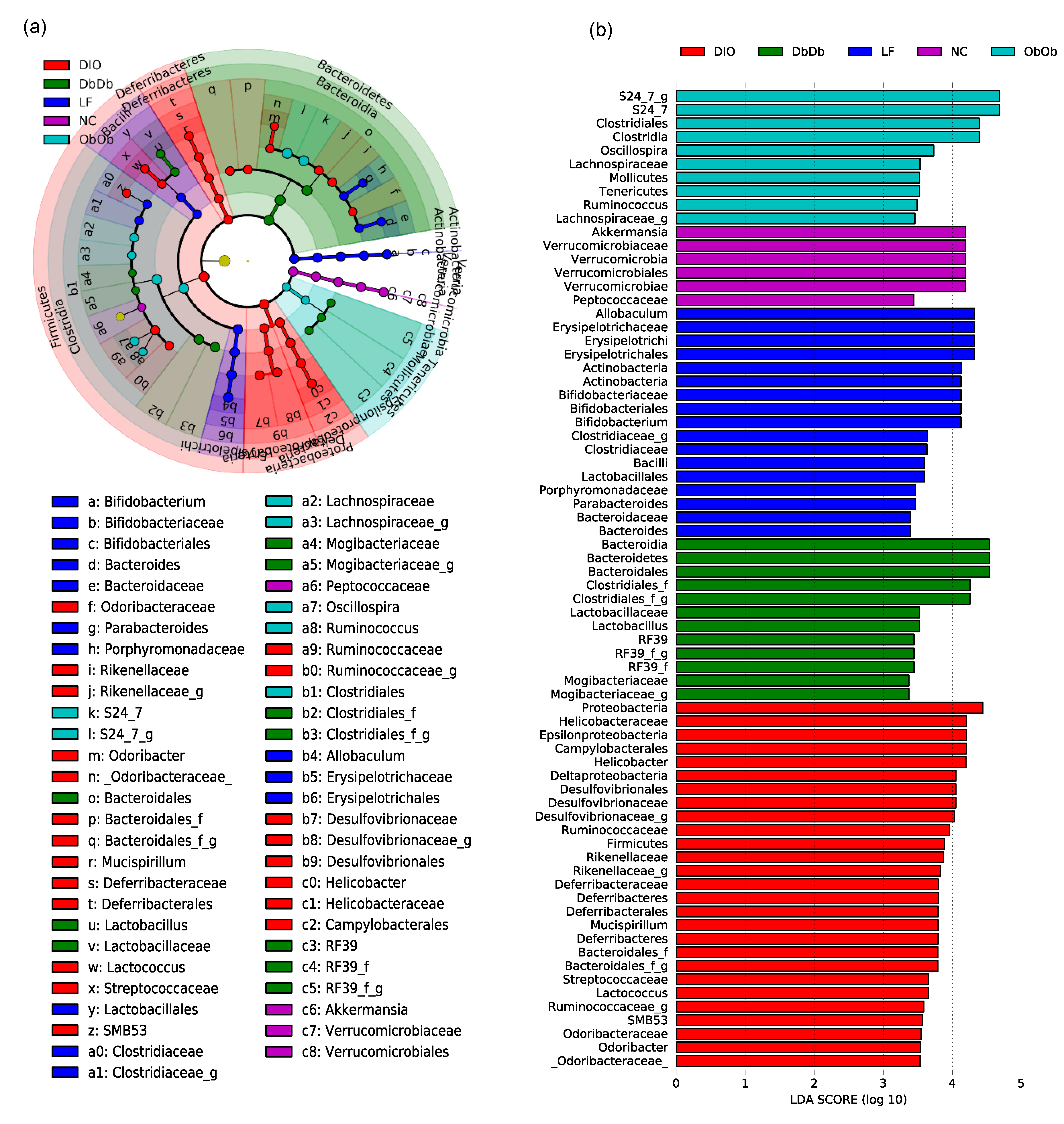
2.1. Diet-Induced and Genetically Induced Obese Mice Harbor Distinct Gut Microbiomes
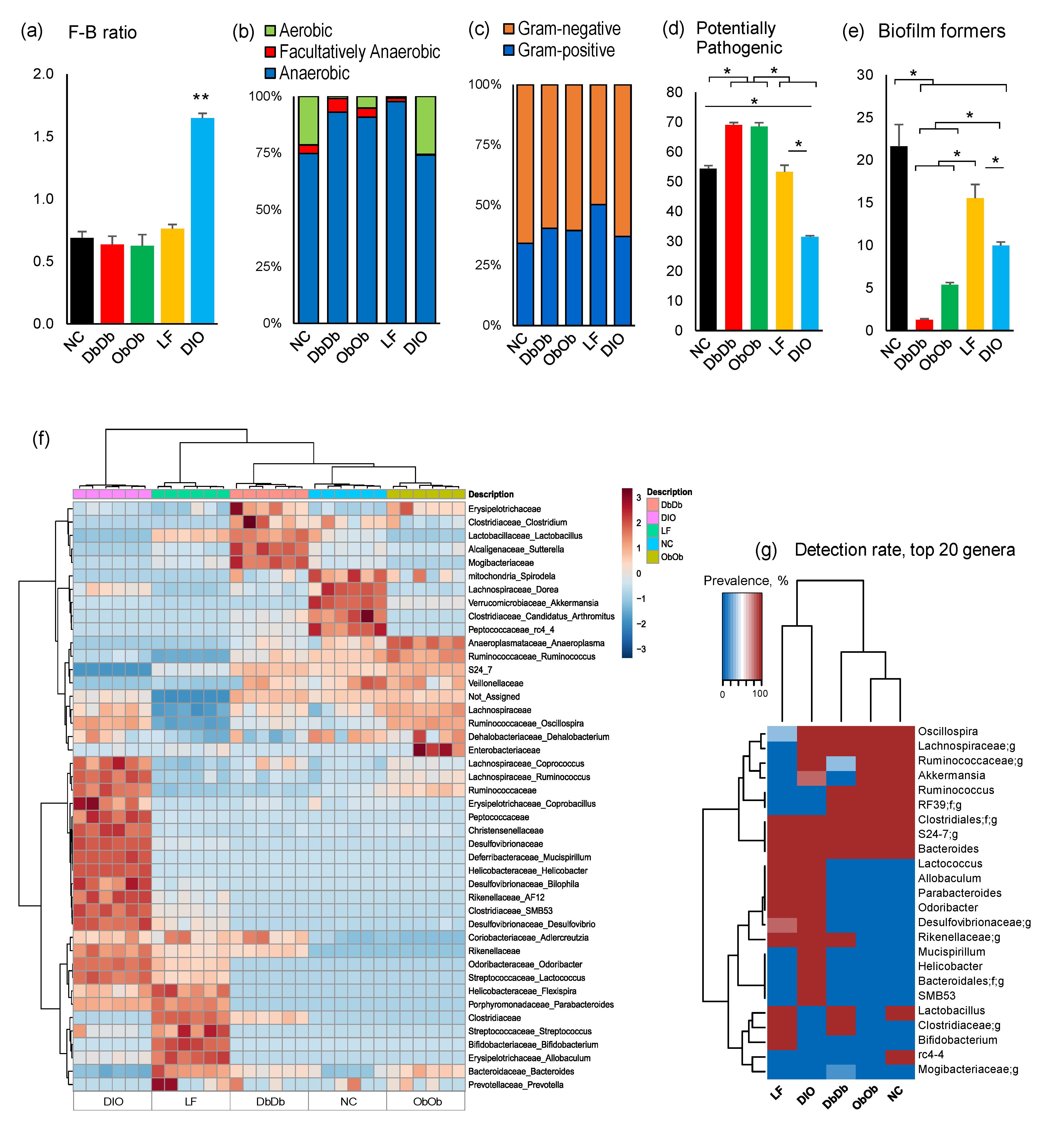
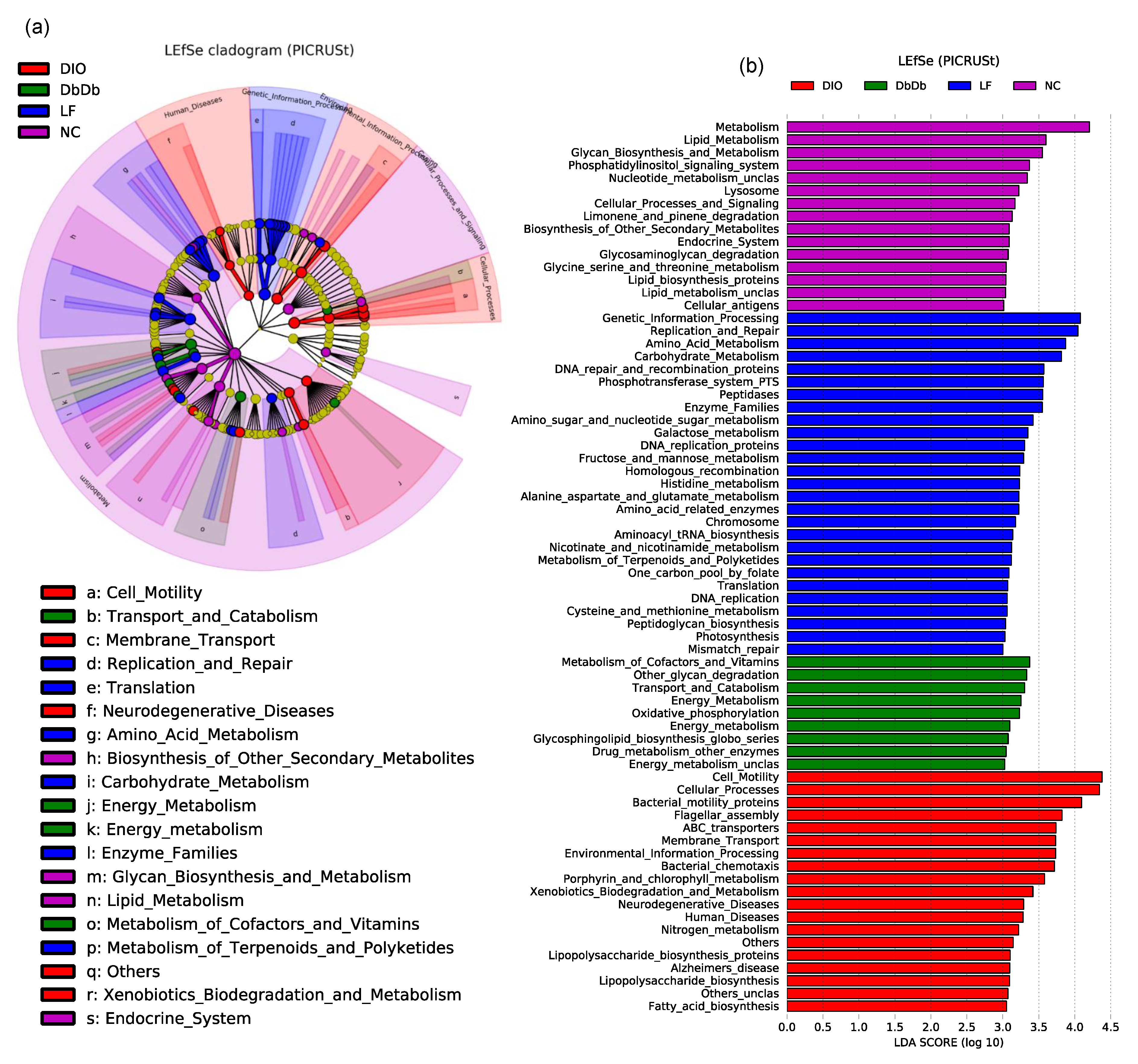
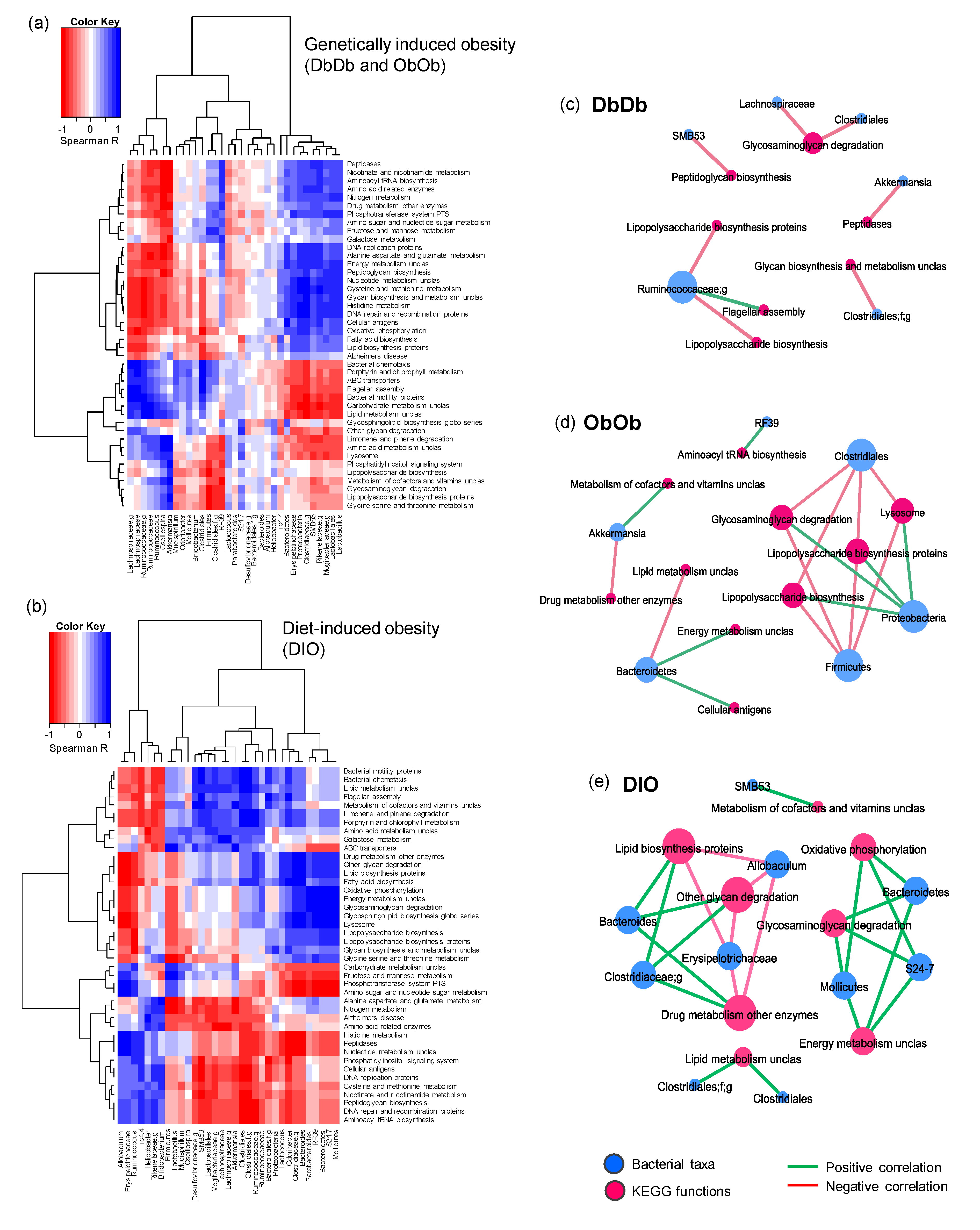
2.2. Diet-Induced Versus Genetically Induced Obese Mice Demonstrate Unique Signatures of Gut Microbiome Community Structure and Function
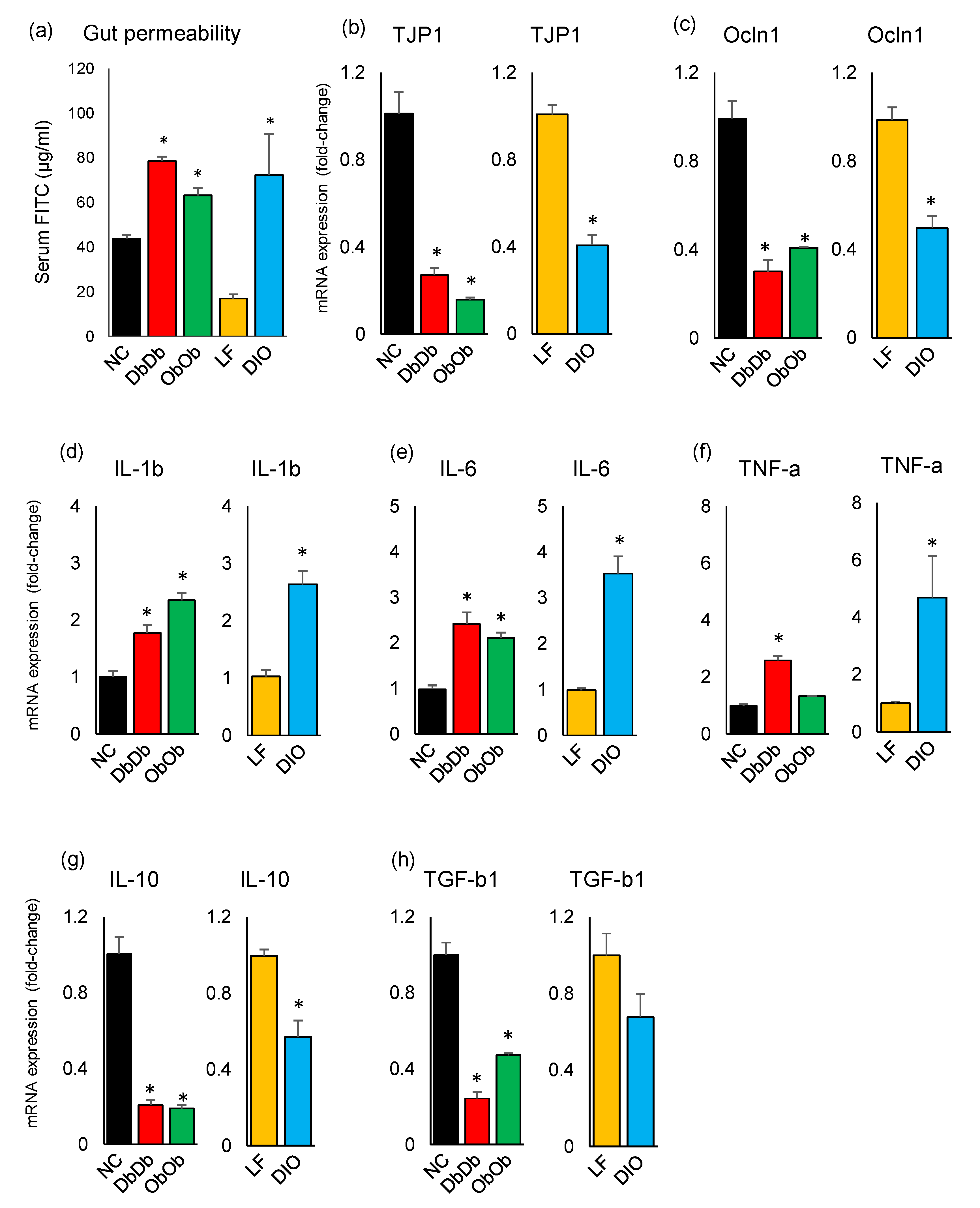
2.3. Diet-Induced and Genetically Induced Obese Mice Exhibit Higher Intestinal Permeability and Inflammation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Gut Microbiome Analysis
4.3. Gut Permeability Measurement
4.4. Real-Time PCR Assays
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DIO | Diet-induced obesity |
| ObOb | Leptin-deficient (Lepob/ob) |
| DbDb | Leptin-receptor-deficient (Leprdb/db) |
| LF | Low-fat |
| NC | Normal chow |
| HFD | High-fat diet |
| T2D | Type-2 diabetes |
| LDA | Linear discriminatory analysis |
| LEfSe | Linear discriminatory analysis effect size |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOs | KEGG orthologs |
| QIIME | Quantitative Insights into Microbial Ecology |
| PICRUSt | Phylogenetic Investigation of Communities by Reconstruction of Unobserved States |
| FITC | Fluorescein isothiocyanate |
| TJP | Tight-junction proteins |
| IL | Interleukins |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| PCoA | Principal coordinate analysis |
| EMP | Earth Microbiome Project |
References
- Boulange, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, L.B.; Ruhlemann, M.C.; Koch, M.; Fuqua, B.; Laucke, G.; Boehm, R.; Bang, C.; Franzosa, E.A.; Hubenthal, M.; Rahnavard, A.; et al. Obese individuals with and without type 2 diabetes show different gut microbial functional capacity and composition. Cell Host Microbe 2019, 26, 252–264.e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; Francois, P.; de Vos, W.M.; et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajala, M.W.; Patterson, C.M.; Opp, J.S.; Foltin, S.K.; Young, V.B.; Myers, M.G., Jr. Leptin acts independently of food intake to modulate gut microbial composition in male mice. Endocrinol. 2014, 155, 748–757. [Google Scholar] [CrossRef] [Green Version]
- Pfalzer, A.C.; Nesbeth, P.D.; Parnell, L.D.; Iyer, L.K.; Liu, Z.; Kane, A.V.; Chen, C.Y.; Tai, A.K.; Bowman, T.A.; Obin, M.S.; et al. Diet- and genetically-induced obesity differentially affect the fecal microbiome and metabolome in Apc1638N mice. PLoS ONE 2015, 10, e0135758. [Google Scholar] [CrossRef] [Green Version]
- Ellekilde, M.; Krych, L.; Hansen, C.H.; Hufeldt, M.R.; Dahl, K.; Hansen, L.H.; Sorensen, S.J.; Vogensen, F.K.; Nielsen, D.S.; Hansen, A.K. Characterization of the gut microbiota in leptin deficient obese mice - Correlation to inflammatory and diabetic parameters. Res. Vet. Sci. 2014, 96, 241–250. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Xie, H.; Wen, Z.; Zhang, Y.; Wu, C.; Huang, L.; Wu, J.; Xie, C.; Wang, T.; et al. Gut microbiota composition and structure of the Ob/Ob and Db/Db mice. Int. J. Endocrinol. 2019, 2019, 1394097. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mazcorro, J.F.; Ivanov, I.; Mills, D.A.; Noratto, G. Influence of whole-wheat consumption on fecal microbial community structure of obese diabetic mice. Peer J. 2016, 4, e1702. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A.; et al. High-fat diet alters gut microbiota physiology in mice. Isme J. 2014, 8, 295–308. [Google Scholar] [CrossRef]
- Naito, Y.; Uchiyama, K.; Takagi, T. A next-generation beneficial microbe: Akkermansia muciniphila. J. Clin. Biochem. Nutr. 2018, 63, 33–35. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Roopchand, D.E.; Carmody, R.N.; Kuhn, P.; Moskal, K.; Rojas-Silva, P.; Turnbaugh, P.J.; Raskin, I. Dietary polyphenols promote growth of the gut bacterium akkermansia muciniphila and attenuate high-fat diet-induced metabolic syndrome. Diabetes 2015, 64, 2847–2858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Qin, Q.; Liu, M.; Zhang, X.; He, F.; Wang, G. Akkermansia muciniphila can reduce the damage of gluco/lipotoxicity, oxidative stress and inflammation, and normalize intestine microbiota in streptozotocin-induced diabetic rats. Pathog. Dis. 2018, 76, fty028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Yadav, H. Bacterial translocation from the gut to the distant organs: An overview. Ann. Nutr. Metab. 2017, 71, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, R.; Shively, C.A.; Appt, S.A.; Register, T.C.; Michalson, K.T.; Vitolins, M.Z.; Yadav, H. Gut microbiome composition in non-human primates consuming a western or mediterranean diet. Front Nutr. 2018, 5, 28. [Google Scholar] [CrossRef]
- Nagpal, R.; Wang, S.; Ahmadi, S.; Hayes, J.; Gagliano, J.; Subashchandrabose, S.; Kitzman, D.W.; Becton, T.; Read, R.; Yadav, H. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci. Rep. 2018, 8, 12649. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, S.; Nagpal, R.; Wang, S.; Gagliano, J.; Kitzman, D.W.; Soleimanian-Zad, S.; Sheikh-Zeinoddin, M.; Read, R.; Yadav, H. Prebiotics from acorn and sago prevent high-fat-diet-induced insulin resistance via microbiome-gut-brain axis modulation. J. Nutr. Biochem. 2019, 67, 1–13. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Isme J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Razazan, A.; Nagpal, R.; Jain, S.; Wang, B.; Mishra, S.P.; Wang, S.; Justice, J.; Ding, J.; McClain, D.A.; et al. Metformin reduces aging-related leaky gut and improves cognitive function by beneficially modulating gut microbiome/goblet cell/mucin axis. J. Gerontol. A. Biol. Sci. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Wang, S.; Ahmadi, S.; Nagpal, R.; Jain, S.; Mishra, S.P.; Kavanagh, K.; Zhu, X.; Wang, Z.; McClain, D.A.; Kritchevsky, S.B.; et al. Lipoteichoic acid from the cell wall of a heat killed Lactobacillus paracasei D3-5 ameliorates aging-related leaky gut, inflammation and improves physical and cognitive functions: From C. elegans to mice. Geroscience 2020, 42, 333–352. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic. Acids. Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagpal, R.; Mishra, S.P.; Yadav, H. Unique Gut Microbiome Signatures Depict Diet-Versus Genetically Induced Obesity in Mice. Int. J. Mol. Sci. 2020, 21, 3434. https://doi.org/10.3390/ijms21103434
Nagpal R, Mishra SP, Yadav H. Unique Gut Microbiome Signatures Depict Diet-Versus Genetically Induced Obesity in Mice. International Journal of Molecular Sciences. 2020; 21(10):3434. https://doi.org/10.3390/ijms21103434
Chicago/Turabian StyleNagpal, Ravinder, Sidharth P Mishra, and Hariom Yadav. 2020. "Unique Gut Microbiome Signatures Depict Diet-Versus Genetically Induced Obesity in Mice" International Journal of Molecular Sciences 21, no. 10: 3434. https://doi.org/10.3390/ijms21103434
APA StyleNagpal, R., Mishra, S. P., & Yadav, H. (2020). Unique Gut Microbiome Signatures Depict Diet-Versus Genetically Induced Obesity in Mice. International Journal of Molecular Sciences, 21(10), 3434. https://doi.org/10.3390/ijms21103434