1. Introduction
Ageing-related cognitive disorders, including age-associated memory impairment (AAMI), mild cognitive impairment (MCI) and senile dementia, have become common health threats to the elderly population. MCI manifests as an intermediate condition between age-related cognitive decline and dementia and represents a prodromal stage before the development of Alzheimer’s disease (AD) [
1,
2]. AD is one of the most common neurodegenerative disorders, characterised by a progressive memory decline, cognitive dysfunctions, amyloid β1-42 (Aβ) plaque accumulation, neurofibrillary tangle formation and occurrence of neurogenic/inflammatory responses in the central nervous system (CNS) [
3].
Of interest, over the last years, alterations of the enteric bacteria-neuro-immune network have been proposed to be involved in the onset of AD and related gut dysfunctions [
4]. In particular, changes in gut microbiota composition and impairments of the intestinal epithelial barrier (IEB) could determine the accumulation of enteric Aβ and phosphorylated tau (p-tau) proteins, which, in turn, could trigger enteric and peripheral neurogenic/inflammatory responses, and contribute to bowel motor disturbances as well as CNS neuroinflammation and neurodegeneration via gut–brain ascending pathways [
4,
5]. Indeed, both pre-clinical and human studies have shown that AD is associated with several changes in gut microbiota composition, signs of enteric inflammation as well as colonic accumulation of Aβ and p-tau tangle-like structures, which could lead to intestinal motor dysfunctions [
6,
7,
8,
9]. In particular, transgenic AD mice were found to display gut dysbiosis, intestinal Aβ and amyloid protein precursor (APP) accumulation, activation of intestinal inflammatory pathways, neuronal loss and enteric glial activation in the early stages of AD [
10,
11]. Other authors reported rearrangements of enteric neuronal coding, characterised by a decrease in neuronal nitric oxide synthase (nNOS) and choline acetyltransferase (ChAT) in mice with early AD [
12]. However, whether colonic dysmotility, enteric AD-related protein accumulation and colonic inflammation represent the earliest events in AD, occurring in the prodromal stage of the disease, presently remains unclear. Likewise, the relationship between onset of cognitive impairment and bowel inflammation, colonic dysmotility and AD-related proteins before the full development of brain pathology remains to be explored in depth.
The present study was designed to examine the relationship between the onset of cognitive deficiencies and colonic dysmotility/inflammation, enteric depositions of AD-related proteins (Aβ, tau, p-tau, α-synuclein (α-syn) and their heterocomplexes) in the prodromal phases of AD in the SAMP8 spontaneous AD model. The SAMP8 mouse is one of the accelerated senescence strains that develop spontaneously early learning and memory deficiencies, with similar features to those observed in AD [
13,
14]. It is an excellent model for studying age-dependent cognitive decline associated with MCI and the subsequent development of AD [
15,
16]. Indeed, SAMP8 mice develop early learning and memory deficiencies since their young age (3–5 months), progressing then toward the development of full AD pathology (8–12 months), where they display the main pathophysiological and clinical features of AD, including the deposition of Aβ 1–40 or 1–42 proteins in hippocampal granules; hyperphosphorylation of tau protein; increase in α-syn, presenilin, oxidative damage, glutamate and nNOS levels; and decrease in ChAT activity [
14]. Thus, owing to these features, the SAMP8 mouse can be regarded as an extremely valuable model for investigating intestinal symptoms and alterations in the prodromal stages of AD [
14,
17,
18,
19,
20]. On these bases, since our intent was to examine whether the onset of cognitive impairment was associated with the occurrence of intestinal symptoms, we decided to perform a timing at four, six and eight months of age in SAMP8 mice to examine the onset of intestinal dysfunctions in the earliest stages of AD (MCI) before the full development of brain pathology. Gaining knowledge in this setting is critical for a better understanding of the mechanisms underlying bowel dysfunctions in AD and designing rational therapeutic approaches.
3. Discussion
Several lines of evidence suggest that, in the early stages of AD, changes in gut microbiota composition, impaired IEB, AD-related protein accumulation in intestinal tissues and enteric inflammation could contribute to CNS pathology and related intestinal dysfunctions [
4]. However, current evidence does not allow establishing clear relationships among colonic dysmotility; enteric AD-related protein accumulation, including Aβ, p-tau, α-syn and their heterocomplexes; bowel inflammation; and AD pathology since the earliest stages of the disease. In this context, our purpose was to examine the occurrence of bowel dysmotility, enteric AD-related protein accumulation, colonic inflammation and IEB impairment in a murine model of accelerated senescence (SAMP8 mouse) in the early phase of AD, preceding the full development of brain pathology. The SAMP8 mouse develops spontaneously early learning and memory deficits, with similar features to those observed in AD patients. Of interest, the SAMP8 mouse, which develops a severe disease after eight months of age, can be an extremely valuable model to investigate intestinal symptoms in the prodromal stage of AD [
13,
14,
25].
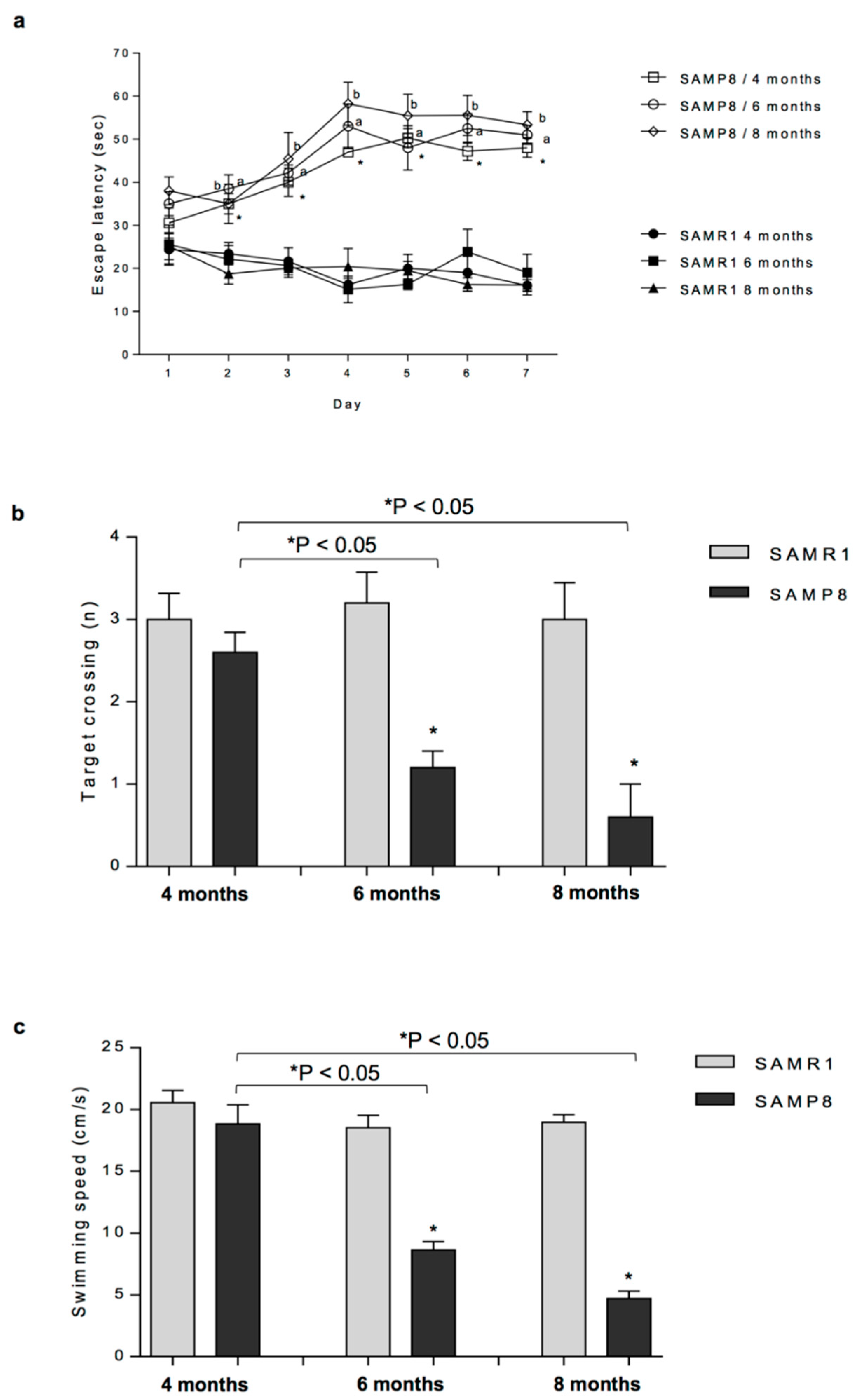
As a first step, we attempted to determine a possible relationship between the onset of cognitive and motor impairments and colonic dysmotility in SAMP8 mice. To pursue this goal, we performed the MWM test and evaluated the faecal output in SAMP8 and SAMR1 animals at four, six and eight months of age. Our results show that SAMP8 mice displayed an impairment of cognitive and motor functions along with a significant decrease in faecal output starting from six months of age, thus indicating that, in SAMP8 animals, the alterations of colonic motility appear in the prodromal phase of AD, before the full development of CNS pathology. Consistently with our findings, several studies have shown that both patients with MCI and early AD are characterised by infrequent bowel movements and constipation [
26,
27].
Based on results from in vivo experiments, we decided to focus the attention on SAMP8 animals at six months of age, in order to examine the mechanisms underlying the intestinal motor dysfunctions associated with the onset of the cognitive and motor symptoms, before the full development of AD. Therefore, in the second part of the study, to verify whether the impaired in vivo colonic motility in SAMP8 mice at six months of age might result from changes in enteric neurotransmission, we examined the patterns of in vitro excitatory (cholinergic and tachykininergic) and inhibitory (nitrergic) motor pathways. Our results show that electrically evoked cholinergic and tachykininergic contractions of colonic muscle preparations from SAMP8 mice were significantly decreased, while electrically evoked inhibitory nitrergic responses were unchanged, resulting in an impaired overall colonic propulsive motility. These findings support the view that the cognitive decline is associated with altered excitatory control of colonic motility, thus providing new insights into the pathophysiological mechanisms underlying the occurrence of bowel dysfunctions in the prodromal stages of AD. Subsequently, to verify whether the decrease in cholinergic and tachykininergic colonic contractions could depend on changes in the density of muscarinic or NK1 tachykininergic receptors on smooth muscle cells, we examined the myogenic colonic contractions through direct stimulation of muscarinic and NK1 receptors with carbachol and exogenous substance P, respectively. Our results show no changes in colonic myogenic cholinergic and NK1-mediated tachykininergic contractions, thus suggesting that the impairments of colonic excitatory contractile responses could be ascribed to an impairment of both cholinergic and tachykininergic excitatory neurotransmission.
Taken together, our results provide the first demonstration that rearrangements of enteric excitatory neuronal motor pathways could contribute to colonic dysmotility occurring in SAMP8 mice in the prodromal stages of AD. Of note, these results are in line with a recent study showing that APP/presenilin 1 (PS1) transgenic AD mice displayed a remodelling of enteric neuronal coding, characterised by a decrease in nNOS and ChAT [
12]. However, these authors evaluated the density of nitrergic and cholinergic neurons, omitting the assessment of colonic in vitro motor activity.
Of note, several lines of evidence suggest that enteric AD-related protein accumulation, including Aβ and p-tau proteins, shape the immune/inflammatory responses that, in turn, could contribute to bowel motor dysfunctions since the earliest stages of AD [
5]. Indeed, enteric Aβ and p-tau protein aggregates and increased faecal calprotectin levels have been observed in AD patients at different stages of the disease [
6,
7].
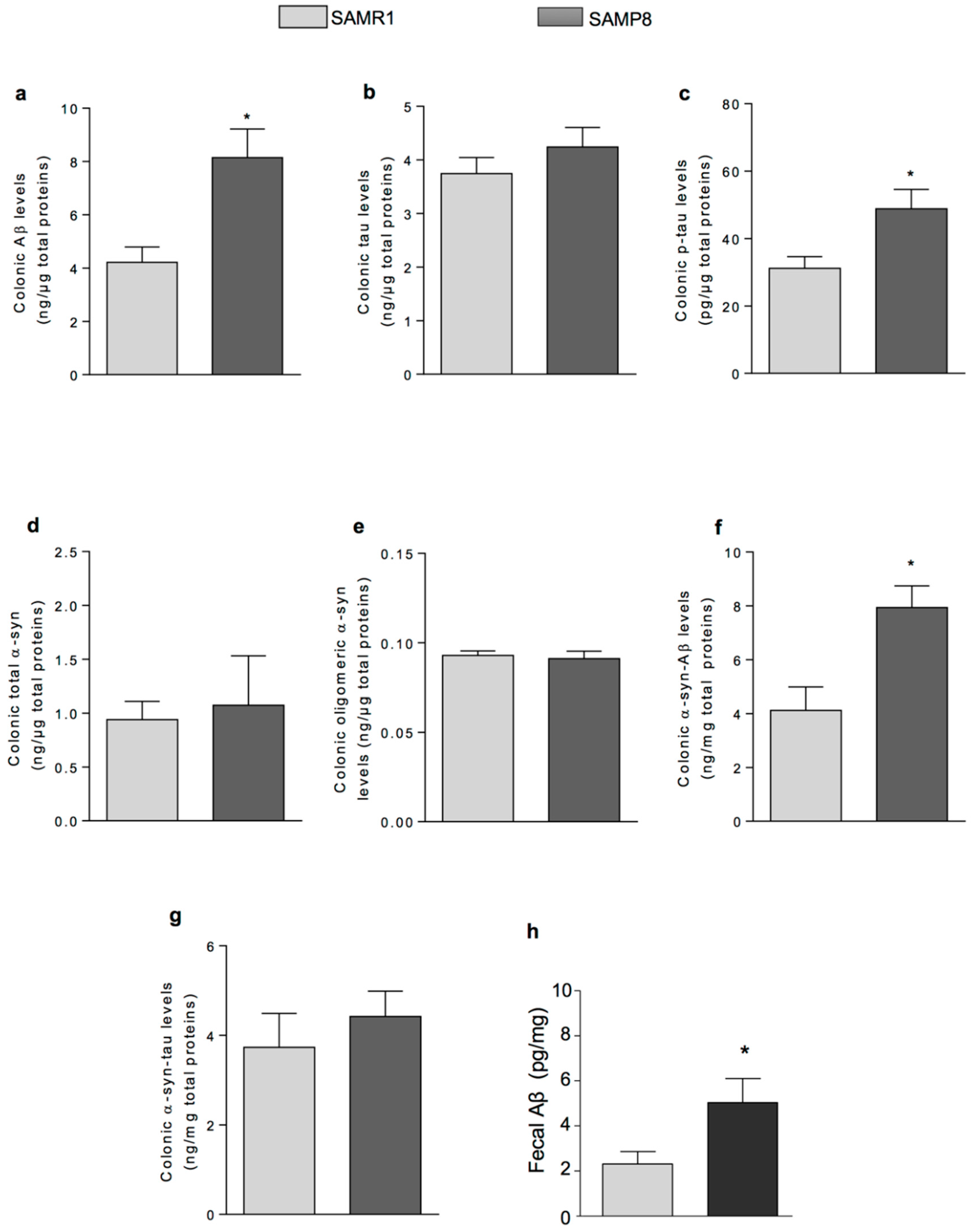
Based on the above knowledge, in the third part of the present study, we focused our attention on investigating the presence of AD-related proteins (i.e., α-syn, tau and Aβ) and their heteroaggregates, including α-syn-tau and α-syn-Aβ, as well as inflammation in colonic tissues from SAMP8 mice. In addition, we evaluated the faecal levels of Aβ which is the dominant variant of Aβ deposits in the human brain [
28]. SAMP8 mice displayed an increase in faecal Aβ as well as colonic Aβ and p-tau levels, as compared with controls. These findings reflect Aβ and p-tau accumulation detected in brain tissues from SAMP8 at six months in previous studies, and suggest that AD-related protein deposits in intestinal tissues correlate with those detected in the brain from SAMP8 mice at early stages of the disease [
25,
29]. By contrast, no variation of colonic total and oligomeric α-syn levels were found when comparing SAMP8 and SAMR1 animals. These data suggest that α-syn is not involved in the enteric AD-protein accumulation in the SAMP8 model. With regard to the assessment of AD-related protein heterocomplexes, we found an increase in the levels of α-syn-Aβ and α-syn-tau deposits in colonic tissues from SAMP8, suggesting that enteric Aβ and tau accumulation could increase their interaction with α-syn. These results are in line with our previous findings showing increased levels of α-syn-Aβ and α-syn-tau heterocomplexes in the brain and red blood cells from SAMP8 mice at six months [
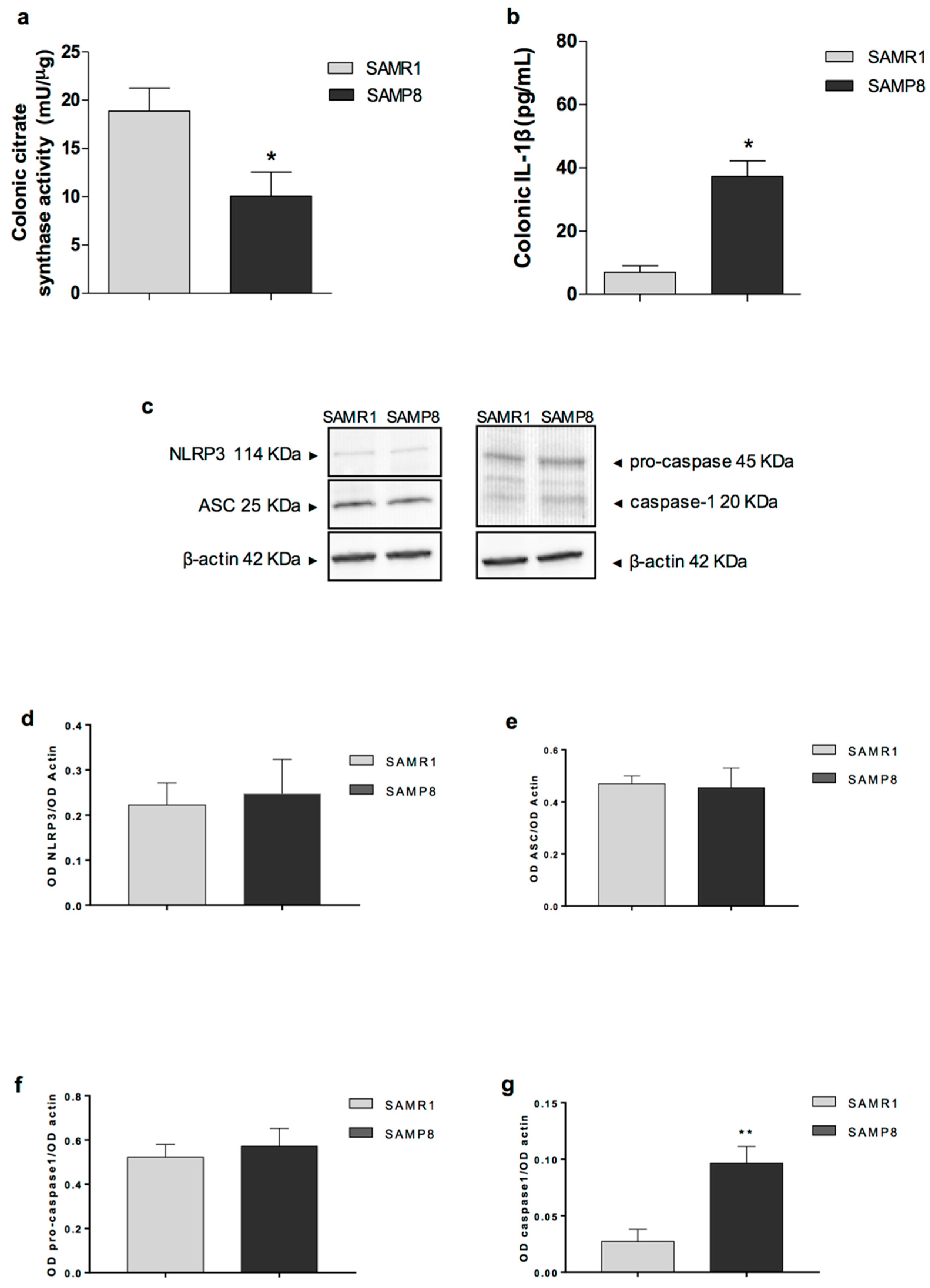
25]. Moreover, we observed an increase in IL-1β levels in colonic tissues from SAMP8 animals, suggesting the occurrence of inflammatory responses in the large bowel of early AD mice. Overall, these observations suggest that, in early AD, the accumulation of AD-related proteins and their heterocomplexes could promote neurogenic/inflammatory responses, which, in turn, could contribute to bowel motor disturbances.
Of note, the accumulation of AD-related proteins and their heteroaggregates could promote mitochondrial dysfunctions (a hallmark of Aβ-induced neuronal toxicity in AD) in the colon of SAMP8 mice [
30]. Therefore, we went on to evaluate the citrate synthase activity, referred to as a suitable marker of mitochondrial activity, which decreases dramatically in several organs and tissues during ageing [
31], in colonic tissues from SAMP8 animals. This rate-limiting mitochondrial enzyme is involved in the first step of the Krebs cycle and catalyses the condensation reaction of the acetate residue from acetyl coenzyme A and oxalacetate to form citrate in mitochondria [
32]. In our study, the citrate synthase activity was significantly decreased in colonic tissues from SAMP8 animals at six months. Such a decrease could involve different intestinal cell types, including intestinal epithelial cells (i.e., Lgr5+ crypt based columnar cells characterised by high basal mitochondrial activity) [
33,
34] and intestinal innate immune/inflammatory cells, including macrophages, regarded as immune sentinels which sense several pathological stimuli, including Aβ, that, in turn, is known to activate inflammatory pathways and mitochondrial dysfunction [
35,
36].
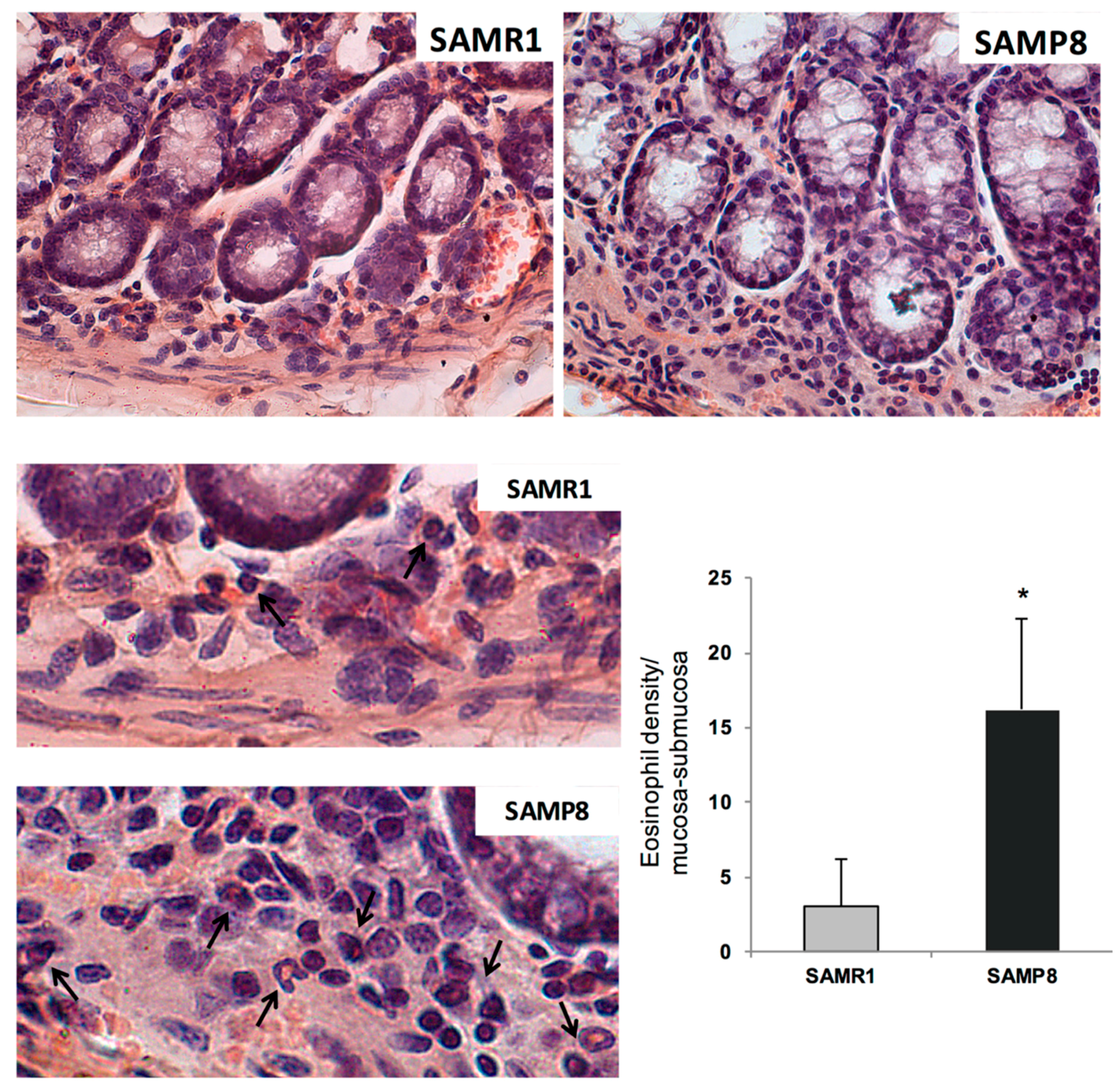
These findings suggest that enteric AD protein accumulation, colonic inflammation, and mitochondrial dysfunction are among the earliest events in AD that could contribute to bowel dysmotility. The presence of bowel inflammation in SAMP8 mice was further corroborated by an increase in eosinophil within the colonic tunica mucosa and submucosa. These results are in line with previous studies, showing an increase in Aβ and p-tau protein expression, as well as immune/inflammatory cell activation in intestinal tissues from TgCRND8 and APP/PS1 mice (genetic models of AD) since the early stages of AD [
11,
37].
It is noteworthy that the accumulation of Aβ proteins, mitochondrial dysfunction and the increase in IL-1β levels in colonic tissues from SAMP8 mice suggest the involvement of the NLRP3 inflammasome multiprotein complex in the onset of enteric inflammation [
21]. In this respect, we evaluated the expression of inflammasome components, including NLRP3, ASC, and caspase-1 in colonic tissues from SAMP8 mice, and we found a significant increase in caspase-1 cleavage, while no changes in NLRP3 and ASC expression were detected. The increase in caspase-1 cleavage, along with the increase in Aβ proteins, mitochondrial dysfunction, and IL-1β levels, indicate the activation of the well-established pattern of canonical inflammasome pathway [
22]. Interestingly, a similar picture has been observed in brain tissues from AD mice, where NLRP3 activation and Aβ protein deposition, mitochondrial dysfunction, and IL-1β were detected [
38].
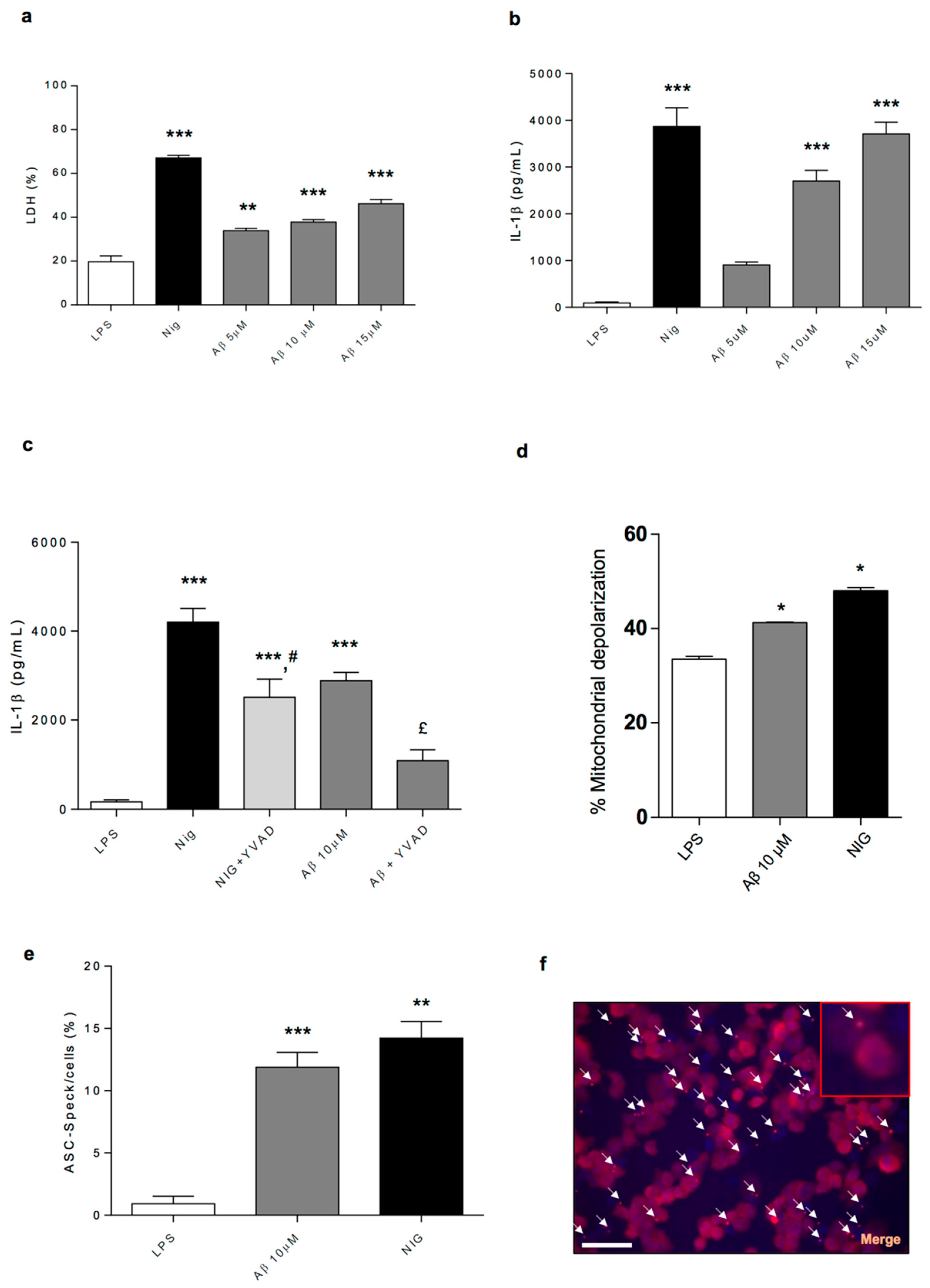
Based on the present findings, it is conceivable that, in early AD, the accumulation of Aβ proteins promotes mitochondrial dysfunction and inflammasome activation, which, in turn, shaping neurogenic/inflammatory responses, could contribute to bowel dysmotility. In support of this view, the accumulation of Aβ proteins in AD has been proposed to contribute to mitochondrial dysfunction as well as to activate the NLRP3 inflammasome complex in the CNS [
39]. In an attempt of confirming this hypothesis, in the fourth part of the present study, we went on to characterise in vitro the molecular mechanisms through which Aβ, regarded as the dominant variant of AD-related proteins, can determine NLRP3 inflammasome activation and mitochondrial dysfunction in immune cells. To pursue this goal, we performed experiments in the LPS-primed PMA-differentiated THP-1 cell line, an established model to investigate monocyte/macrophage functions. In particular, we tested the ability of Aβ of inducing the release of IL-1β through direct activation of NLRP3 inflammasome and to alter mitochondrial functions. Interestingly, our results show that Aβ was able to stimulate NLRP3 activation and induce IL-1β release in a concentration-dependent fashion in THP-1 cells through the stimulation of ASC oligomerisation, a pivotal step in NLRP3 activation. In support of these findings, Aβ did not induce the release of IL-1β in ASC
-/- THP-1 cells or in WT cells pre-treated with a caspase-1 inhibitor. These findings corroborate previous findings indicating that Aβ accumulation promotes the release of IL-1β through the direct activation of the NLRP3 inflammasome complex in immune cells [
39]. In addition, the Aβ treatment of THP-1 cells decreased the mitochondrial potential, thus suggesting that Aβ is involved also in the mechanisms leading to alteration of mitochondrial activity.
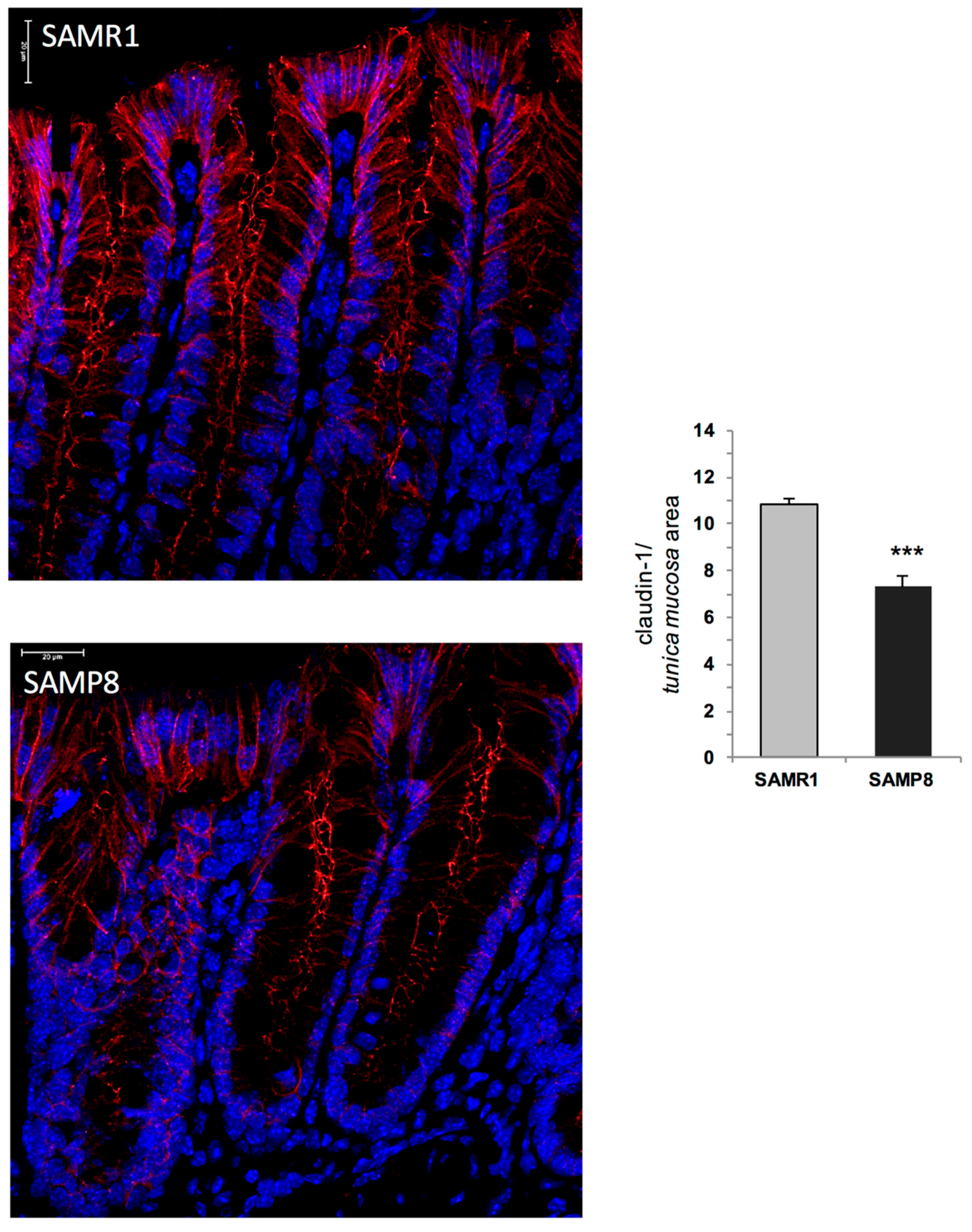
Based on the above findings, it is conceivable that, in early AD mice, the enteric accumulation of AD-related proteins, with particular regard for Aβ, determines NLRP3 activation and mitochondrial dysfunctions, which, in turn, promote the occurrence of enteric neurogenic/inflammatory conditions that could contribute to alterations of bowel motility. However, whether the Aβ-induced NLRP3 activation, besides shaping the immune/inflammatory responses, contributes also to alter the enteric neuronal pathways, or whether both events occur concomitantly, remains to be clarified. In addition, studies aimed at evaluating the specific enteric neuronal and immune/inflammatory cell types (i.e., myenteric neurons, enteric glial cells and macrophages) involved in the onset of enteric inflammation associated with AD would be required. The occurrence of enteric inflammation, besides contributing to bowel motor dysfunctions, could alter the IEB, with consequent alterations of enteric permeability [
40]. Therefore, in the last part of the study, we evaluated the morphological changes of the intestinal mucosal barrier in SAMP8 mice. In particular, we focused our attention on claudin-1 protein, a pivotal tight junction component involved in the maintenance of IEB integrity [
41]. Our results showed a decreased expression of claudin-1 in SAMP8 mice, suggesting an impairment of IEB in early AD animals. Such a decrease could contribute to bacterial translocation into the intestinal mucosa, with consequent chronicisation of the ongoing inflammatory response and further worsening of bowel motor dysfunctions [
42]. A similar trend has been observed in patients with early Parkinson’s disease, characterised by alterations of occludin and zonulin-1 expression in colonic tissues [
43,
44]. Based on these findings, it is conceivable that the enteric inflammation might impair the IEB integrity, with consequent alterations of intestinal permeability, and further worsening of bowel motor dysfunctions. However, the correlation among enteric inflammation, impaired IEB, and bowel dysmotility requires confirmation by means of additional experimental approaches. In addition, since the NLRP3 inflammasome is expressed also in intestinal epithelial cells [
22], further experiments aimed at evaluating whether Aβ protein can promote inflammasome activation in intestinal epithelial cells should be implemented.
4. Materials and Methods
4.1. Animals
SAMP8 (Senescence-Accelerated Mouse-Prone 8) mice (2 months old, 20–25 g body weight), a spontaneous genetic model of AD, and their control strain SAMR1 (Senescence-Accelerated Mouse-Resistant 1) (2 months old, 20–25 g body weight) were purchased from ENVIGO Srl (San Pietro al Natisone UD, Italy) and employed throughout the study.
The animals were fed with standard laboratory chow and tap water ad libitum, and were not employed for at least 1 week after their delivery to the laboratory. They were housed, one in a cage, in temperature-controlled rooms on a 12-h light cycle at 22–24 °C and 50–60% humidity. Standard diet (Altromin International, Germany; SD, TD.2018) provided 3.1 kcal/g, of which 18% as fats, 24% as proteins and 58% as carbohydrates. The feeding behaviour (frequency and amount) was assessed until the day before the sacrifice. At the end of study, animals were euthanised. Animals were housed, three in a cage, in temperature-controlled rooms on a 12-h light cycle at 22–24 °C and 50–60% humidity. Their care and handling were in accordance with the provisions of the European Community Council Directive 210/63/UE, recognised and adopted by the Italian Government. The experiments were approved by the Ethical Committee for Animal Experimentation of the University of Pisa and by the Italian Ministry of Health on february 25th 2016 (Authorisation No. 198/2016-PR).
The SAMP8 mouse is one of the accelerated senescence strains that develops spontaneously early learning and memory deficits, with similar features to those observed in AD [
13,
14]. Of note, the SAMP8 mouse displays the main clinical and pathophysiological features to those observed in AD patients, including Aβ 1–40 or 1–42 proteins in hippocampal granules, hyperphosphorylation of tau protein, increase in α-syn, presenilin, oxidative damage, glutamate levels, and nNOS, along with the decrease in ChAT activity [
14].
SAMP8 and SAMR1 animals at four, six and eight months of age were subjected to Morris Water Maze (MWM) test in order to evaluate alterations of cognitive functions from the initial phases of early learning and memory deficiencies until the full development of AD. The day after the evaluation of cognitive and motor performances, animals were employed for the assessment of faecal output, as described below. One hour after the evaluation of faecal output, SAMP8 and SAMR1 animals at six months of age were euthanised and colonic specimens were dissected and processed for functional experiments and other assays, as described below.
4.2. Evaluation of Cognitive Functions
MWM test: The MWM uses a round pool (90 cm in diameter and 60 cm in height) filled with opaque water (26 ± 1 °C temperature). The pool was divided into four quadrants of equal area, designated arbitrarily as northeast (NE), southeast (SE), southwest (SW) and northwest (NW). A circular platform (10 cm diameter and 30 cm height) was placed in the centre of one quadrant. All external clues were constant for the spatial orientation of mice. A camera mounted directly above the centre of the round pool monitored animal movements. The camera image was digitalised and fed to a computerised tracking system that monitored and stored animal movements [
45].
The MWM test consists of visible-platform acquisition training, hidden-platform training, and probe trial as previously reported [
25]. The platform was in the same location for both visible-platform training and hidden platform training. In the acquisition training, the escape latency was assessed for each animal (time required to reach the platform). Mice were placed on the platform for 15 s before being released into the water. Mice were allowed to swim and find the visible platform within 60 s. Each animal was subjected to sessions of four trials every day for 2 days. After the daily trial, mice were returned to their home cages for resting. In the hidden-platform training, performed by submerging the platform 1.5 cm below the surface of the water, escape latency was evaluated over the next 5 days. Each animal was subjected to sessions of four trials every day. Finally, on the eighth day, the platform was removed from the tank for the probe trial. The number of target crossings the number of entries into the target quadrant, the time spent in the target quadrant where the platform was placed, the swimming speed and swim distance were assessed in 60 s. Data were expressed as raw values.
4.3. Evaluation of Faecal Output
Faecal output was recorded from 9:00 AM to 10:00 AM on each day. Each animal was removed from its home cage and placed in a clean plastic cage without food or water for 1 h. Stools were collected immediately after expulsion and placed in sealed tubes. At the end of the trial, the stools were counted and weighed (total weight), then dried overnight at 65 °C and weighed again to estimate the dry weight.
4.4. Recording of Colonic Contractile Activity
The contractile activity of colonic muscle preparations was recorded as previously described by [
46]. Following euthanasia, the abdomen was immediately opened, the colon was removed and placed in Krebs solution. Segments of the colon were opened along the mesenteric insertion and cut along the longitudinal axis into strips of approximately 3 mm in width and 8 mm in length. The preparations were set up in organ baths containing Krebs solution at 37 °C, bubbled with 95% O
2 + 5% CO
2 and connected to isometric transducers (constant load = 0.5 g). Krebs solution had the following composition (mM): NaCl 113, KCl 4.7, CaCl
2 2.5, KH
2PO
4 1.2, MgSO
4 1.2, NaHCO
3 25 and glucose 11.5 (pH 7.4 ± 0.1). The mechanical activity was recorded by BIOPAC MP150 (Biomedica Mangoni, Pisa, Italy). Each preparation was allowed to equilibrate for at least 30 min, with intervening washings at 10 min intervals. A pair of coaxial platinum electrodes was positioned at a distance of 10 mm from the longitudinal axis of each preparation to deliver electrical stimulation by a BM-ST6 stimulator (Biomedica Mangoni, Pisa, Italy). Electrical stimuli (ES) were applied: 10-s single trains consisting of square wave pulses (0.5 ms, 30 mA). At the end of the equilibration period, each preparation was repeatedly challenged with ES, and the experiments started when reproducible responses were obtained (usually after two or three stimulations). The tension developed by each preparation was normalised by the wet tissue weight and expressed as grams per gram of wet tissue (g/g tissue).
Preliminary experiments were performed to select the appropriate ES frequency, as well as carbachol (CCh) or exogenous substance P (SP) concentration, which elicited submaximal contractions. These preliminary experiments allowed to select the frequency of 10 Hz and the concentration of 10 μM of CCh and 1 μM of SP.
4.5. Design of Experiments on Colonic Contractions
In the first series of experiments, electrically induced contractions were recorded from colonic preparations maintained in standard Krebs solution.
In the second set of experiments, electrically induced contractions were recorded from colonic preparations maintained in Krebs solution containing guanethidine (adrenergic blocker 10 μM), N-acetyl-l-tryptophan 3,5-bis(trifluoromethyl) benzylester (L-732,138, neurokinin NK1 receptor antagonist, 10 μM), 5-fluoro-3-[2-[4-methoxy-4-[[(R)-phenylsulphinyl]methyl]-1-piperidinyl]ethyl]-1H-indole (GR159897, NK2 receptor antagonist, 1 μM), (R)-[[(2-phenyl-4-quinolinyl)carbonyl]amino]-methyl ester benzeneacetic acid (SB218795, NK3 receptor antagonist, 1 μM) and atropine (muscarinic receptor antagonist, 1 μM) in order to evaluate the patterns of colonic contractions driven by nitrergic pathways.
In the third series, colonic specimens were maintained in Krebs solution containing Nω-nitro- L-arginine methylester (L-NAME, nitric oxide synthase inhibitor, 100 μM), guanethidine, L-732,138, GR159897 and SB218795 and contractions were elicited by ES in order to examine the patterns driven by excitatory cholinergic nerves.
The fourth series of experiments was designed to study the neurogenic NK1-mediated contribution to muscle contraction. For this purpose, colonic tissues were maintained in Krebs solution containing L-NAME, guanethidine, atropine sulphate, GR159897 and SB218795 and electrically evoked motor responses were recorded.
In the fifth set of experiments, colonic cholinergic contractions were evoked by direct pharmacological activation of muscarinic receptors located on smooth muscle cells. For this purpose, colonic preparations were maintained in Krebs solution containing tetrodotoxin (TTX, 1 μM) and stimulated with CCh (10 μM).
In the last series, tachykininergic NK1-mediated contractions were evoked by direct pharmacological activation of NK1 receptors located on smooth muscle cells. For this purpose, colonic specimens were maintained in standard Krebs solution, added with TTX and stimulated with exogenous SP (1 μM).
4.6. Collection of the Colon and Total Protein Quantification
Phosphate buffered saline (PBS) was added to colonic tissue specimens, and the samples were sonicated. A Bradford assay was performed to quantify the total proteins present in the samples. Then, sodium dodecyl sulphate (SDS) was added to samples to achieve a concentration of 100 μg/100 μL of total proteins.
4.7. Preparation of Oligomeric α-Syn and α-Syn Biotinylated Antibody
Recombinant α-syn was incubated in parafilm sealed tubes at 37 °C for 4 days in an Eppendorf Thermomixer under continuous mixing (1000 rpm) [
47]. A reaction among Sulpho-NHS-LC-Biotin (Pierce, Rockford, IL, USA) (200 mg) and the 211 mouse monoclonal antibody (mAb) (Santa Cruz Biotechnology, USA) was allowed to prepare α-syn biotinylated antibody [
48]. Then, the mixture was desalted on Bio-Spin-6 columns (BIO-RAD, Langford, UK) to eliminate excess uncoupled biotin [
49,
50,
51].
4.8. Evaluation of Tissue and Faecal AD-related Protein Levels and Their Heterocomplexes
Colonic total α-syn: Colonic total α-syn was evaluated by a “home-made” sandwich enzyme-linked immunosorbent assay (ELISA) system [
49,
52]. First, 60 μL/well of full length rabbit polyclonal antibody to α-syn (SC-10717, Santa Cruz Biotechnology, with epitope mapping amino acids 672-714 of Aβ, i.e., Aβ 1–42), diluted 1:100 in poly-L-ornithine (diluted in 50 mM NaHCO
3 pH 9.6), were used to coat wells, which were then incubated overnight at 4 °C. After washes, 200-μL/well of bovine serum albumin (BSA) 1% were added to each well and incubated for 1 h at 37 °C to block non-specific sites. Colonic samples were added to each well (10 μg/100 μL) and incubated at 25 °C for 2 h. As primary antibody, 75 μL/well of a mouse monoclonal antibody to α-syn (SC-12767, Santa Cruz Biotechnology, with epitope mapping the carboxy-terminus of Aβ), diluted 1:200 in PBS-BSA-Triton, were employed and incubated at 37 °C for 2 h. Then, 100 μL/well of an anti-mouse-horseradish peroxidase (HRP) antibody, diluted 1:2000 in PBS-BSA-Triton, were used as detection antibody, and incubated at 37 °C for 1.5 h [
49]. Next, 100 µL of the chromogenic substrate (3,3′,5,5′-tetramethylbenzidine, TMB, Thermo Scientific) were added to each well. The absorbance was read at 450 nm after the addition of 100 μL of the stop solution (0.4 N HCl) in each well. All measurements were made in duplicate to achieve a minimal inter-assay variability. A recombinant human α-syn solution at different concentrations, diluted in PBS, was used to build the standard curve for ELISA assay [
49,
50,
51,
53].
Colonic oligomeric α-syn: Colonic oligomeric α-syn was evaluated by an immunoenzymatic assay, as described previously [
47,
50]. A mouse monoclonal antibody to α-syn (SC-12767, Santa Cruz Biotechnology) was used to pre-coat wells, and it was left in incubation overnight at room temperature. BSA 1% (200 μL/well) was added for 1 h at 37 °C. The colonic tissue (0.4 μg/100 μL) was introduced into each well and incubated for 2 h at 25 °C. To detect α-syn oligomers, an α-syn biotinylated antibody (that binds human α-syn on the amino acid residues 121–125) (75 μL/well) was employed as primary antibody. A streptavidin HRP conjugate antibody (1:1000, GE Healthcare) was used as detection antibody (100 μL/well). TMB was added to each well (100 μL/well) and colouring was monitored. Then, stop solution (100 μL/well) was added to block the colorimetric reaction. The absorbance was read at 450 nm.
The levels of the interaction oligomeric α-syn in the samples was quantified through a standard curve that was built using different concentrations of recombinant human oligomeric α-syn (Human Alpha Synuclein oligomer ELISA kit, MBS730762, MyBioSource) [
49].
Colonic total Aβ: The amount of Aβ in colonic tissue was determined through immuno-enzymatic assay, as described previously [
48,
49]. A specific rabbit polyclonal antibody to Aβ (SC-9129, Santa Cruz Biotechnology), diluted 1:100 in poli-L-ornithine, was added (60 μL) to each well and maintained overnight at 4 °C. To block aspecific sites, BSA 1% (200 μL/well) was added for 2 h at 37 °C. The colonic tissue (0.25 μg/100 μL) was introduced into each well and incubated at 25 °C for 1 h. A goat polyclonal antibody to Aβ (SC-5399, Santa Cruz Biotechnology) (75 μL/well) was employed and incubated for 1,5 h at 25 °C; then, a donkey anti-goat-HRP antibody (Santa Cruz Biotechnology), diluted 1:2500 in PBS-BSA-Triton, was used against the primary antibody and incubated at 37 °C for 1 h [
49]. Conclusively, the wells were incubated with TMB (100 μL/well). After adding the stop solution (100 μL/well), the absorbance was read at 450 nm [
49,
54].
Faecal total Aβ: Aβ levels in the stools were measured by an ELISA kit (KMB3441, Invitrogen), as previously described [
54]. For this purpose, faecal samples (30 mg), stored previously at −80 °C, were weighed, thawed and homogenised in 0.4 mL of 5 M guanidine-HCl/50 mM Tris (pH 8.0 at room temperature) for 3−4 h. Stools were diluted ten-fold with cold PBS with 1× protease inhibitor cocktail (Sigma), centrifuged at 16,000×
g for 20 min at 4 °C, and then the supernatants were transferred into clean microcentrifuge tubes and kept on ice. Subsequently, a protease inhibitor cocktail with a serine protease inhibitor 1 mM (AEBSF, Sigma) was added since serine proteases can rapidly degrade Aβ peptides. Aliquots (100 μL) of supernatants were then used for the assay. Faecal Aβ levels were expressed as picogram per milligram of feces.
Colonic total tau: The quantification of total tau in colonic tissue was assessed by an immuno-enzymatic assay, as described previously [
48,
49]. A specific mouse monoclonal antibody to tau (SC-32274, Santa Cruz Biotechnology, with epitope mapping the carboxyl-terminus of tau protein), diluted 1:100 in poly-L-ornithine, was used (60 μL/well) to pre-coat the plate and incubated overnight at 4 °C. BSA 1% (200 μL/well) was added for 1 h at 37 °C, and later on, the colonic tissue (2 μg/100 μL) was incubated for 2 h at 25 °C. A rabbit polyclonal antibody to tau (SC-5587, Santa Cruz Biotechnology) (75 μL/well), diluted 1:250 in PBS-BSA-Triton, was incubated for 2 h at 37 °C. Successively, a goat anti-rabbit-HRP antibody (Invitrogen), diluted 1:2000 in PBS-BSA-Triton, was incubated for 1.5 h. The TMB was added to each well (100 μL/well) and the absorbance was read at 450 nm after adding stop solution (100 μL/well) [
49,
55].
Colonic p-tau: p-tau levels in colonic tissue were measured through an immuno-enzymatic assay, as described previously [
49]. A specific antibody to tau (SC-32274, Santa Cruz Biotechnology), diluted 1:100 in poly-L-ornithine, was used (60 μL/well) to pre-coat the plate and left overnight at 4 °C. BSA 1% (200 μL/well) was added for 2 h at 37 °C. The colonic tissue (1 μg/100 μL) was incubated for 2 h at 25 °C. A polyclonal antibody to tau (70R-32555, Fitzgerald, detecting endogenous levels of tau only when phosphorylated at Thr181) (75 μL/well), diluted 1:5000 in PBS-BSA-Triton, was incubated for 1.5 h at 37 °C. Then, an HRP antibody (Invitrogen), diluted 1:2000 in PBS-BSA-Triton, was incubated for 1.5 h. After the incubation with TMB (100 μL/well), the absorbance was measured at 450 nm after adding the stop solution (100 μL/well) [
49].
Colonic α-syn-Aβ heterocomplexes: To quantify the interactions of α-syn with Aβ in colonic tissue, a “home-made” sandwich ELISA system was developed [
49,
56].
The levels of the interaction between α-syn and Aβ in the samples was quantified through a standard curve [
49], which was built using different concentrations of recombinant human α-syn and recombinant human Aβ. The solution was prepared by incubating in parafilm-sealed tubes 1 mg of each protein, in 2 mM SDS, and maintaining it at 37 °C for 36 h in an “Eppendorf Thermomixer” with continuous mixing (500 rpm) [
49,
57]. All measurements were performed in duplicate to reduce inter-assay variability. Sixty microlitres/well of a rabbit polyclonal antibody to Aβ (SC-9129, Santa Cruz Biotechnology), diluted 1:100 in poly-L-ornithine, were used to pre-coat wells and incubated overnight at room temperature. The colonic tissue (1 μg/100 μL) was added to each well and incubated for 2 h at 25 °C. Two hundred microlitres of BSA 1% were added to each well for 20 min at 37 °C, to block aspecific sites. Seventy-five microlitres of mouse monoclonal anti-α-syn antibody (SC-12767, Santa Cruz Biotechnology), diluted 1:200 in Milk 5%, were employed and incubated at 37 °C for 2 h. Subsequently, 100 µL of goat anti-mouse-HRP antibody (Santa Cruz), diluted 1:2000 in Milk 5%, were incubated for 1.5 h at 37 °C [
49]. Then, 100 µL/well of TMB were added. Absorbance was evaluated at 450 nm after adding 100 μL/well of stop solution.
Colonic α-syn-tau heterocomplexes: To detect quantitatively the interactions of α-syn with tau in colonic tissue, a “home-made” sandwich ELISA was developed [
49,
50,
51,
56]. The levels of the interaction between α-syn and tau in tissue samples was quantified through a standard curve [
49], which was built using different concentrations of recombinant human α-syn and recombinant human tau. The solution was prepared by incubating in parafilm-sealed tubes 1 mg of each protein, diluted in 2 mM SDS, and maintaining it at 37 °C for 1 h in an Eppendorf Thermomixer with continuous mixing (500 rpm) [
49,
50,
51]. All measurements were performed in duplicate to lower inter-assay variability. A pre-coating of the ELISA plate was carried out using a goat polyclonal α/β-syn antibody (SC-7012, Santa Cruz Biotechnology), diluted 1:100 in poly-L-ornithine, left overnight at room temperature. After incubation of the colonic samples (1 μg/100 μL) in each well for 2 h at 25 °C, BSA 1% (200 μL/well) was added for 20 min at 37 °C to block aspecific sites. As primary antibody, a rabbit polyclonal anti-tau antibody (SC-5587, Santa Cruz Biotechnology), diluted 1:200, was employed for capturing at 37 °C for 2 h. Subsequently, a goat anti-rabbit-HRP antibody was used, as detection antibody, at 37 °C for 1.5 h [
49]. One hundred microlitres/well of TMB were added in each well and the colour was allowed to develop for 30 min at room temperature. Absorbance was measured at 450 nm after adding 100 μL/well of stop solution.
4.9. Evaluation of Tissues IL-1β Levels
Interleukin (IL)-1β levels in colonic tissues were measured by ELISA kit (R&D system), as described previously [
58,
59]. For this purpose, colonic tissue samples, stored previously at −80 °C, were weighed, thawed and homogenised in 0.4 mL of PBS, pH 7.2/20 mg of tissue at 4 °C, and centrifuged at 10,000×
g for 5 min. Aliquots (100 μL) of supernatants were then used for the assay. Tissue IL-1β levels were expressed as picogram per millilitre.
4.10. Evaluation of Tissue Citrate Synthase Activity
Frozen colonic tissues were homogenised on ice with an ultra-turrax homogeniser (IKA-Werke GmbH & Co., Germany) in a cold buffer (composition: sucrose 250 mM, Tris 5 mM, EGTA 1 mM, Triton X-100 0.02%, pH 7.4). Then, homogenates were centrifuged at 12,000× g for 15 min at 4 °C (EuroClone, Speed Master 14 R centrifuge, Italy). The supernatant was used for determination of the citrate synthase activity, and the protein concentration in the supernatant was determined spectrophotometrically by Bradford assay (Bio-Rad, USA), using a microplate reader (EnSpire, PerkinElmer, USA). Then, proteins were diluted in Tris-buffer 100 mM (pH 8.2) containing 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB, 100 µM) and acetyl-coenzyme A (100 µM). The assay was performed in 96-well plates (1 µg of proteins per well) and the reaction was initiated by addition of oxalacetate 500 µM. The absorption of the reaction product was measured spectrophotometrically at 30 °C and 412 nm every 30 s for 15 min. Citrate synthase activity was determined by comparing the activity in the samples to that of known concentrations of the isolated enzyme (Sigma-Aldrich, St. Louis, MI, USA). Citrate synthase activity was expressed in mU/µg protein. Data were analysed by a computer fitting procedure (software: GraphPad Prism 5.0).
4.11. Western Blot Analysis of NLRP3, ASC and Caspase-1 Expression
The colonic tissues were weighed and homogenised in lysis buffer, using a polytron homogeniser, as described by Richter et al. [
60]. Proteins were quantified with the Bradford assay. Proteins (30 μg) were separated onto a pre-cast 4-20% polyacrylamide gel (Mini-PROTEAN
® TGX gel, Biorad) and transferred to PVDF membranes (Trans-Blot
® TurboTM PVDF Transfer packs, Biorad). Membranes were blocked with 3% BSA diluted in Tris-buffered saline (TBS, 20 mM Tris-HCl, pH 7.5, 150 mM NaCl) with 0.1% Tween 20. Primary antibodies against β-actin (monoclonal, diluted 1:5000, A5316, Sigma Aldrich), nucleotide-binding oligomerisation domain leucine rich repeat and pyrin domain containing protein 3 (NLRP3) (polyclonal, diluted 1:1000, ab214185, Abcam), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) (monoclonal, diluted 1:1000, D2W8U, Cell Signaling) and caspase-1 (polyclonal, diluted 1:1000, ab1872, Abcam) were used. Secondary antibodies were obtained from Abcam (anti-mouse ab97040, Abcam, and anti-rabbit ab6721, Abcam). Protein bands were detected with ECL reagents (Clarity™ Western ECL Blotting Substrate, Biorad). Densitometry was performed by ImageJ software.
4.12. Histological Evaluation of Eosinophils
Sections from formalin-fixed full-thickness colonic samples were processed for routine (haematoxylin and eosin) and histochemical staining (0.1% toluidine blue in 30% ethanol for 15 min) in order to detect eosinophils, which appeared as deep violet cells. The density of eosinophils was assessed in the tunica mucosa/submucosa. Cells were counted in three different sections from each mouse, and at least 20 randomly selected microscopic fields were examined in each section (objective, 40×). Values obtained from all the examined fields for each rat were averaged and expressed as cell number per square millimetre of tunica mucosa/submucosa areas, which were estimated by the Image Analysis System “L.A.S. software v.4.” These values were then used to calculate mean values for each experimental group.
4.13. Immunohistochemistry of Claudin-1
Sections (epithelial cells) were incubated overnight at 4 °C with the primary anti-claudin-1 antibody. Sections were then exposed to appropriate biotinylated immunoglobulins, peroxidase-labelled streptavidin complex and 3.3′-diaminobenzidine tetrahydrochloride, counterstained and examined by a Leica DMRB light microscope equipped with a DFC480 digital camera (Leica Microsystems, Cambridge, UK). For each group, representative photomicrographs were shot and analysed quantitatively using the Image Analysis System “L.A.S. software version 4.5” (Leica Microsystems, Cambridge, UK). Two blind investigators (C.I. and C.S.) carried out cell-counting independently and assessed the colorimetric threshold values to detect antigen expression levels. Immunostaining expression was calculated as ratio between area of the stained fields and the total tissue area examined (percentage positive pixels (PPP)), as previously reported [
58,
61]. Data obtained from all the examined fields for each rat were averaged and used to calculate mean values ± SEM for each experimental group, which were plotted in graphs.
4.14. In Vitro Assays on NLRP3 Inflammasome
Wilde-type (WT) and ASC knock out (ASC-/-) human monocytic cell lines (THP-1) were donated by Prof. Veit Hornung (Ludwig Maximilian University of Munich) and cultured in RPMI 1640 media (Sigma) supplemented with 10% FBS (PAA Laboratories), 100 units/mL penicillin, and 100 µg/mL streptomycin (Sigma). Cells were plated in 24-well plates at a density of 5 × 105 cells/well and treated with phorbol 12-myristate 13-acetate (PMA, 0.5 µM). After 3 h, the medium was removed, fresh media was added, and cells were incubated overnight (37 °C, 5% CO2).
In the first series of experiments, cells were lipopolysaccharide (LPS)-primed (1 µg/mL, 4 h) to induce pro-IL-1β expression before treatment with nigericin (a standard NLRP3 inflammasome activator, 10 µM, 1 h) or Aβ (5, 10 and 15 µM, 6 h), as described by [
24,
62,
63].
In the second series of experiments, LPS-primed (1 µg/mL, 4 h) cells were treated for 15 min with vehicle (0.5% dimethyl sulfoxide, DMSO) or caspase-1 inhibitor (YVAD, 100 µM) before the addition of nigericin (10 µM, 1 h) or Aβ (10 µM, 6 h), as described by [
24].
In the third series of experiments, LPS-primed (1 µg/mL, 6 h) ASC-/- THP-1 cells were treated with nigericin (standard NLRP3 inflammasome activator, 10 µM, 1 h) or Aβ (10 µM, 6 h).
WT and ASC-/- THP-1 treated with nigericin or Aβ in the presence or absence of YVAD, respectively, were incubated for 1 h or 6 h, respectively, before the collection of supernatants and the lysis of cells for analysis of IL-1β processing and release. IL-1β in cell supernatants was quantified by an ELISA kit (R&D Systems), following the protocols provided by the manufacturer. IL-1β concentration was expressed as picogram per millilitre.
4.14.1. Cell Death Measurement
Cell death was measured using quantitative assessment of lactate dehydrogenase (LDH) levels in the medium. CytoTox 96® Non-Radioactive Cytotoxicity Assay (G1780, Promega) was used in accordance with manufacturer instructions. Plates were read at 490 nm and results are shown as percentage of LDH release relative to the total cells lysed.
4.14.2. ASC Speck Detection and Quantification
Cells were plated as described above on glass coverslips. THP-1 cells were LPS-primed (1 µg/mL, 4 h) and treated with nigericin (10 µM, 1 h) or Aβ (10 µM, 6 h). Cells were then fixed with 4% paraformaldehyde and 4% sucrose in PBS for 30 min. The cells were permeabilised with 0.1% Triton X-100 and then quenched with 0.25% ammonium chloride. A blocking step for 1 h using 5% BSA, and 5% donkey serum (block solution) was used before incubation with the rabbit anti-ASC (1:500). Coverslips were then washed in PBS. ASC antibodies were detected by incubation with Alexa Fluor 594 conjugated donkey anti-rabbit antibody (1:1000) in blocking solution for 1 h. The coverslips were washed again with PBS and finally in distilled water before being dried and mounted onto a glass slide using ProLong Gold mounting medium containing 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen). Images were taken with an Olympus BX51 upright microscope using a 20×/0.50 Plan Fln objective and captured using a Coolsnap EZ camera (Photometrics) through MetaVue Software (Molecular Devices). To quantify the extent of speck formation, the percentage of cells that contained an ASC speck was counted. Cells from 10 different fields (average of 650 cells/field) were counted for each of the different experiments (
n = 3). Images were analysed using ImageJ (
rsb.info.nih.gov). Data are expressed as the percentage of ASC specks per number of cells per field.
4.14.3. Flow Cytometry Analysis of Mitochondrial Potential
Mitochondrial membrane potential variations are related to apoptotic process, necrotic cell death and caspase-independent cell death. Depolarisation of the inner mitochondrial membrane potential is a reliable indicator of mitochondrial dysfunction and cellular health. Mitochondrial potential was analysed by flow cytometry using the Muse™ MitoPotential Assay Kit from EMD Millipore Bioscience. Mitochondrial potential was evaluated according to the manufacturer’s protocol. Briefly, THP-1 cells were seeded onto 24-well plates at a density of 5 × 104 per well. After 24 h, cells were treated for 6 h with Aβ 10 µM and for 4 h with LPS 1 µg/mL followed by nigericin 2-h treatment at 10 µM, or with no-treatment. After incubation, 1 × 104 cells were resuspended in the 1× Assay Buffer provided. Next, 95 µL of Muse™ MitoPotential working solution were added to 100 μL of cell suspension and incubated at 37 °C for 20 min protected from light. Then, 5 µL of Muse™ 7-AAD working solution were added and incubated at 37 °C for 5 min; cells were analysed by the Muse™ Cell Analyzer.
4.15. Drugs and Reagents
Aβ was purchased by Biochem (Germany). Atropine sulphate, SP, guanethidine monosulphate, nigericin, Ac-YVAD-cmk, bacterial LPS (Escherichia coli 026:B6) and DMSO, sucrose, Tris, EGTA, Triton X-100, Tris-buffer, DTNB, acetil-coenzyme A, oxaloacetate and anti-β-actin-HRP (A3854) were purchased from Sigma Chemicals Co. (St. Louis, MO, USA). Foetal bovine serum (FBS) was obtained from PAA Laboratories. TTX, GR159897, SB218795, L-NAME were obtained from Tocris (Bristol, UK). Anti-IL-1β (anti-human) was purchased by R&D, rabbit anti-ASC (SC-22514-R) by Santa Cruz. Secondary antibody HRP conjugates were from DAKO. Alexa Fluor 594-conjugated donkey anti-rabbit antibody (A-21207) was purchased by Invitrogen.
4.16. Statistical Analysis
The results are presented as mean ± S.E.M. unless otherwise stated. The significance of differences was evaluated by two-way analysis of variance (ANOVA) followed by post hoc analysis with the Fisher LSD test (for paired data), one-way ANOVA followed by post hoc analysis with Bonferroni post hoc test (for paired data) or Student t test (for unpaired data) where appropriate. p values < 0.05 were considered significantly different. All statistical procedures were performed by commercial software (GraphPad Prism, version 7.0 from GraphPad Software Inc., San Diego, CA, USA).
5. Conclusions
The present study provides evidence that, in the SAMP8 AD model, cognitive dysfunctions are associated with enteric AD-related protein accumulation and their heterocomplexes, colonic inflammation, mitochondrial dysfunction, altered IEB and impaired excitatory cholinergic and tachykininergic neurotransmission, which may all contribute to bowel motor dysfunctions since the earliest stages of the disease, before the full development of brain pathology. In this context, inflammasome activation might represent the crossroad between the shaping of enteric neurogenic/inflammatory responses and the onset of bowel motor alterations.
It must be acknowledged, however, that our results do not allow to establish clearly whether the intestinal changes contribute to brain pathology, or whether they occur rather as a consequence of the initiation of central neurodegeneration. In this regard, several lines of evidence support the contention that alterations of the enteric bacteria-neuro-immune network, besides determining intestinal dysfunctions, may contribute also to the pathogenesis of AD [
26,
64,
65]. In particular, changes in gut microbiota composition can promote the pathological accumulation of enteric Aβ protein. Enteric Aβ, regarded also as a prion-like proteinaceous nucleating particle, could then move through myenteric neurons and spread to the CNS, via the neuronal gut–brain axis, contributing directly to the pathogenesis of AD [
66,
67]. In parallel, the enteric Aβ-protein deposition could shape enteric and peripheral neurogenic/inflammatory responses (i.e., activation of NLRP3 inflammasome) and contribute to both bowel motor dysfunctions and neuroinflammation/neurodegeneration in the CNS, via immune gut–brain ascending pathways [
5]. However, whether the Aβ-induced NLRP3 activation, besides shaping immune/inflammatory responses, contributes also to alter the enteric neuronal pathways, or whether both events occur concomitantly, remains to be clarified. In addition, whether the Aβ prion-like protein spreads to the CNS, or whether, through NLRP3 activation, it triggers peripheral and central immune/inflammatory responses, via immune–gut–brain pathways, contributing to brain pathology, remains unclear and deserve further investigations. Moreover, given the relevance of the relationship among gut microbiota, diet and Aβ accumulation, the characterisation of enteric bacteria alterations in SAMP8 mice since the earliest stages of disease, their role in promoting Aβ-protein accumulation, and the impact of diet (i.e., Mediterranean diet, including polyunsaturated fatty acids proteins, vitamins, polyphenols and fibres) in counteracting enteric Aβ-protein accumulation, inflammation, bowel motor symptoms and CNS pathology, remains to be clarified and could represent the logical continuation in this research topic.
Another gap in our study concerns whether the enteric neurogenic/inflammatory alterations in SAMP8 animals could depend on their accelerated ageing, rather than AD-related cognitive impairment. In this regard, we provide evidence that cognitive dysfunctions in SAMP8 animals are associated with enteric AD-related protein accumulation and their heterocomplexes, colonic inflammation, mitochondrial dysfunction, altered IEB and impaired excitatory cholinergic and tachykininergic neurotransmission, which could contribute to bowel motor dysfunctions since the earliest stages of the disease. In addition, in the in vitro experiments, we observed that AD-related proteins, with particular regard for Aβ, promoted inflammasome activation and mitochondrial depolarisation, thus suggesting that the enteric AD-related protein deposition could trigger immune/inflammatory responses and mitochondrial dysfunctions that, in turn, could contribute to bowel dysmotility. Therefore, it is conceivable that the enteric changes observed in SAMP8 mice depend on AD-related protein accumulation in intestinal tissues. However, future investigations in SAMP8 mice at different ages, aimed at evaluating the occurrence of pathological AD-related protein deposition in colonic tissues and bowel dysfunctions, could allow establishing a temporal relationship between intestinal alterations and AD progression.
Overall, these results can provide a basis for better understanding of the mechanisms underlying bowel motor disturbances in AD, thus paving the way to the identification of novel pharmacological approaches to the management of intestinal symptoms associated with AD.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






