The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Intestinal Mucosal Barrier Function
3. Apical Junctional Complex
3.1. Tight Junctions
3.2. Adherens Junctions
4. Defects of the Apical Junctional Complex in Gastrointestinal Disorders
4.1. Inflammatory Bowel Diseases
4.2. Celiac Disease
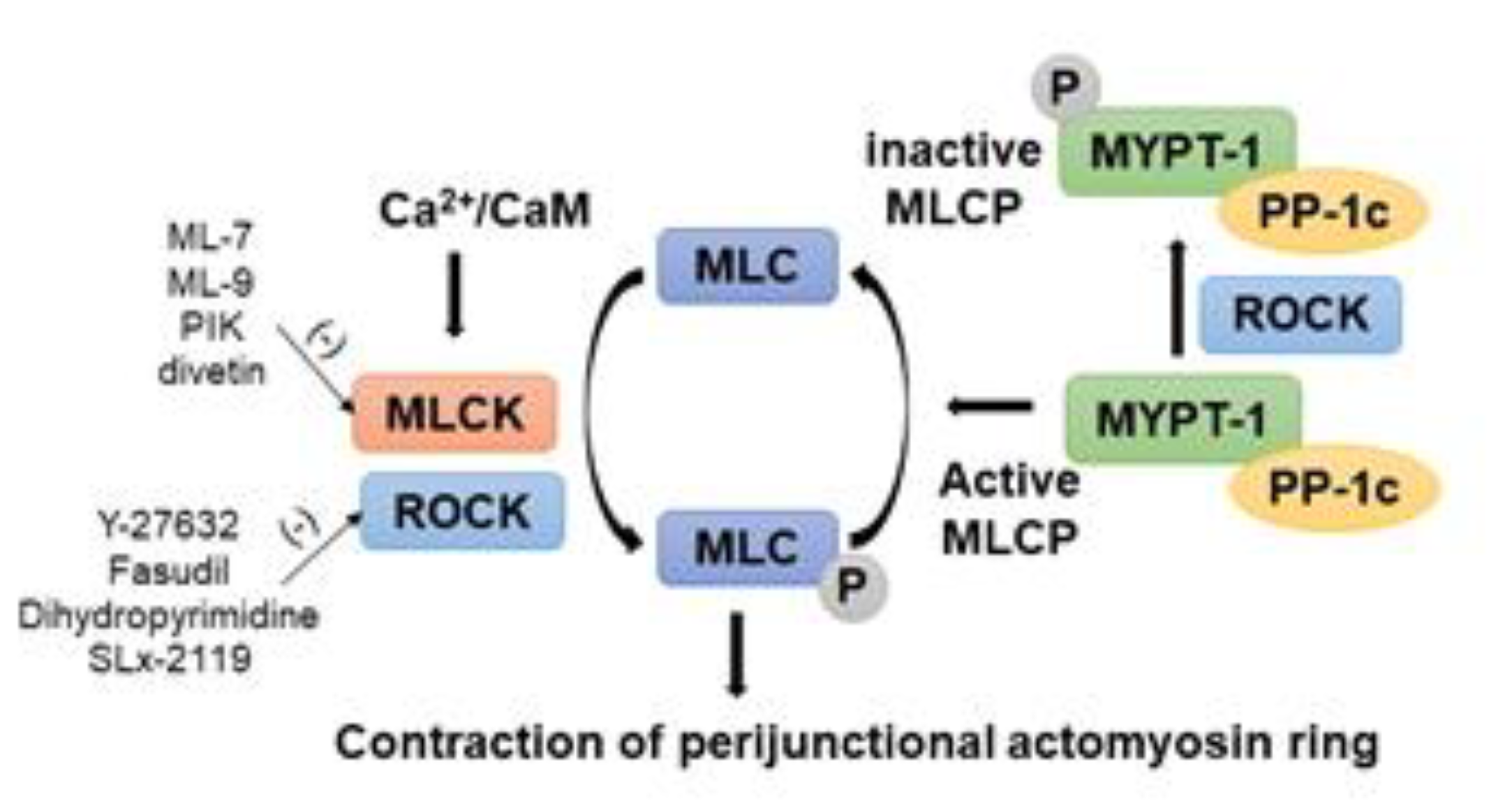
5. Role of MLCK in Phosphorylation of MLC-2 to Regulate Intestinal AJC
6. Role of MLCK in Gastrointestinal Disorders
7. Potential of MLCK Inhibition as a Therapeutic Approach
8. Role of Rho/ROCK Signaling Pathway in Intestinal AJC
9. Role of Rho/ROCK in Digestive Diseases
10. The potential of ROCK Inhibition as a Therapeutic Approach
11. Conclusions
Funding
Conflicts of Interest
References
- Jin, Y.; Blikslager, A.T. ClC-2 regulation of intestinal barrier function: Translation of basic science to therapeutic target. Tissue Barriers 2015, 3, e1105906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blikslager, A.T.; Roberts, M.C.; Rhoads, J.M.; Argenzio, R.A. Prostaglandins I2 and E2 have a synergistic role in rescuing epithelial barrier function in porcine ileum. J. Clin. Investig. 1997, 100, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Gehren, A.S.; Rocha, M.R.; de Souza, W.F.; Morgado-Diaz, J.A. Alterations of the apical junctional complex and actin cytoskeleton and their role in colorectal cancer progression. Tissue Barriers 2015, 3, e1017688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laukoetter, M.G.; Bruewer, M.; Nusrat, A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr. Opin. Gastroenterol. 2006, 22, 85–89. [Google Scholar] [CrossRef]
- Das, P.; Goswami, P.; Das, T.K.; Nag, T.; Sreenivas, V.; Ahuja, V.; Panda, S.K.; Gupta, S.D.; Makharia, G.K. Comparative tight junction protein expressions in colonic Crohn’s disease, ulcerative colitis, and tuberculosis: A new perspective. Virchows Arch. 2012, 460, 261–270. [Google Scholar] [CrossRef]
- Jauregi-Miguel, A.; Fernandez-Jimenez, N.; Irastorza, I.; Plaza-Izurieta, L.; Vitoria, J.C.; Bilbao, J.R. Alteration of tight junction gene expression in celiac disease. J. Pediatr. Gastroenterol. Nutr. 2014, 58, 762–767. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, N.; Chen, Y.; Fu, C.; Huang, K. OTA induces intestinal epithelial barrier dysfunction and tight junction disruption in IPEC-J2 cells through ROS/Ca(2+)-mediated MLCK activation. Environ. Pollut. 2018, 242, 106–112. [Google Scholar] [CrossRef]
- Li, Z.; Gao, M.; Yang, B.; Zhang, H.; Wang, K.; Liu, Z.; Xiao, X.; Yang, M. Naringin attenuates MLC phosphorylation and NF-kappaB activation to protect sepsis-induced intestinal injury via RhoA/ROCK pathway. Biomed. Pharm. 2018, 103, 50–58. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Montcuquet, N.; Roy, M.; Dussaillant, M.; Hugot, J.P.; Barreau, F. Complementary Roles of Nod2 in Hematopoietic and Nonhematopoietic Cells in Preventing Gut Barrier Dysfunction Dependent on MLCK Activity. Inflamm. Bowel Dis. 2017, 23, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Miao, W.; Wu, X.; Wang, K.; Wang, W.; Wang, Y.; Li, Z.; Liu, J.; Li, L.; Peng, L. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCbeta2. Int. J. Mol. Sci. 2016, 17, 1696. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Nalle, S.C.; Shen, L.; Turner, E.S.; Singh, G.; Breskin, L.A.; Khramtsova, E.A.; Khramtsova, G.; Tsai, P.Y.; Fu, Y.X.; et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 2013, 145, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, B.T.; Wang, F.; Shen, L.; Clayburgh, D.R.; Su, L.; Wang, Y.; Fu, Y.X.; Turner, J.R. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 2007, 132, 2383–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Schwarz, B.T.; Graham, W.V.; Wang, Y.; Su, L.; Clayburgh, D.R.; Abraham, C.; Turner, J.R. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology 2006, 131, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R.; Rill, B.K.; Carlson, S.L.; Carnes, D.; Kerner, R.; Mrsny, R.J.; Madara, J.L. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 1997, 273, 1378–1385. [Google Scholar] [CrossRef]
- Zou, Y.; Ma, L.; Zhao, Y.; Zhang, S.; Zhou, C.; Cai, Y. Inhibition of Rho kinase protects against colitis in mice by attenuating intestinal epithelial barrier dysfunction via MLC and the NF-kappaB pathway. Int. J. Mol. Med. 2018, 41, 430–438. [Google Scholar] [CrossRef]
- Watari, A.; Sakamoto, Y.; Hisaie, K.; Iwamoto, K.; Fueta, M.; Yagi, K.; Kondoh, M. Rebeccamycin Attenuates TNF-alpha-Induced Intestinal Epithelial Barrier Dysfunction by Inhibiting Myosin Light Chain Kinase Production. Cell. Physiol. Biochem. 2017, 41, 1924–1934. [Google Scholar] [CrossRef]
- Jin, Y.; Blikslager, A.T. Myosin light chain kinase mediates intestinal barrier dysfunction via occludin endocytosis during anoxia/reoxygenation injury. Am. J. Physiol. Cell Physiol. 2016, 311, 996–1004. [Google Scholar] [CrossRef]
- Choi, W.; Yeruva, S.; Turner, J.R. Contributions of intestinal epithelial barriers to health and disease. Exp. Cell Res. 2017, 358, 71–77. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp. Mol. Med. 2017, 49, e338. [Google Scholar] [CrossRef] [Green Version]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, J.R.; Turner, J.R. Beyond Ussing’s chambers: Contemporary thoughts on integration of transepithelial transport. Am. J. Physiol. Cell Physiol. 2016, 310, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight junction pore and leak pathways: A dynamic duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R.; Buschmann, M.M.; Romero-Calvo, I.; Sailer, A.; Shen, L. The role of molecular remodeling in differential regulation of tight junction permeability. Semin. Cell Dev. Biol. 2014, 36, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Farquhar, M.G.; Palade, G.E. Junctional Complexes in Various Epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [Green Version]
- Staehelin, L.A. Further observations on the fine structure of freeze-cleaved tight junctions. J. Cell Sci. 1973, 13, 763–786. [Google Scholar]
- Yu, D.; Turner, J.R. Stimulus-induced reorganization of tight junction structure: The role of membrane traffic. Biochim. Biophys. Acta 2008, 1778, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin—A Novel Integral Membrane-Protein Localizing at Tight Junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Furuse, M.; Furuse, K.; Sasaki, H.; Tsukita, S.; Tsukita, S. Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J. Cell Biol. 2005, 171, 939–945. [Google Scholar] [CrossRef]
- Martin-Padura, I.; Lostaglio, S.; Schneemann, M.; Williams, L.; Romano, M.; Fruscella, P.; Panzeri, C.; Stoppacciaro, A.; Ruco, L.; Villa, A.; et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J. Cell Biol. 1998, 142, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balda, M.S.; Whitney, J.A.; Flores, C.; Gonzalez, S.; Cereijido, M.; Matter, K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 1996, 134, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.M.; Skare, I.B.; Stankewich, M.C.; Furuse, M.; Tsukita, S.; Rogers, R.A.; Lynch, R.D.; Schneeberger, E.E. Occludin is a functional component of the tight junction. J. Cell Sci. 1996, 109, 2287–2298. [Google Scholar] [PubMed]
- Furuse, M.; Fujimoto, K.; Sato, N.; Hirase, T.; Tsukita, S.; Tsukita, S. Overexpression of occludin, a tight junction-associated integral membrane protein, induces the formation of intracellular multilamellar bodies bearing tight junction-like structures. J. Cell Sci. 1996, 109, 429–435. [Google Scholar]
- Buschmann, M.M.; Shen, L.; Rajapakse, H.; Raleigh, D.R.; Wang, Y.; Wang, Y.; Lingaraju, A.; Zha, J.; Abbott, E.; McAuley, E.M.; et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol. Biol. Cell 2013, 24, 3056–3068. [Google Scholar] [CrossRef]
- Saitou, M.; Fujimoto, K.; Doi, Y.; Itoh, M.; Fujimoto, T.; Furuse, M.; Takano, H.; Noda, T.; Tsukita, S. Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J. Cell Biol. 1998, 141, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [Green Version]
- Kitajiri, S.; Katsuno, T.; Sasaki, H.; Ito, J.; Furuse, M.; Tsukita, S. Deafness in occludin-deficient mice with dislocation of tricellulin and progressive apoptosis of the hair cells. Biol. Open 2014, 3, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 2006, 68, 403–429. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.E.; Daugherty, B.L.; Mitchell, L.A.; Koval, M. Claudins: Control of barrier function and regulation in response to oxidant stress. Antioxid Redox Signal 2011, 15, 1179–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: A lesson from claudin-1-deficient mice. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Tamura, A.; Hayashi, H.; Imasato, M.; Yamazaki, Y.; Hagiwara, A.; Wada, M.; Noda, T.; Watanabe, M.; Suzuki, Y.; Tsukita, S. Loss of claudin-15, but not claudin-2, causes Na+ deficiency and glucose malabsorption in mouse small intestine. Gastroenterology 2011, 140, 913–923. [Google Scholar] [CrossRef]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef]
- Anderson, J.M.; Stevenson, B.R.; Jesaitis, L.A.; Goodenough, D.A.; Mooseker, M.S. Characterization of ZO-1, a protein component of the tight junction from mouse liver and Madin-Darby canine kidney cells. J. Cell Biol. 1988, 106, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: A multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, 1213–1228. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Aponte, A.; Tietgens, A.J.; Gucek, M.; Fredriksson, K.; Anderson, J.M. The N and C termini of ZO-1 are surrounded by distinct proteins and functional protein networks. J. Biol. Chem. 2013, 288, 13775–13788. [Google Scholar] [CrossRef] [Green Version]
- Tsukita, S.; Katsuno, T.; Yamazaki, Y.; Umeda, K.; Tamura, A.; Tsukita, S. Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann. N. Y. Acad. Sci. 2009, 1165, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Umeda, K.; Matsui, T.; Nakayama, M.; Furuse, K.; Sasaki, H.; Furuse, M.; Tsukita, S. Establishment and characterization of cultured epithelial cells lacking expression of ZO-1. J. Biol. Chem. 2004, 279, 44785–44794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooshio, T.; Kobayashi, R.; Ikeda, W.; Miyata, M.; Fukumoto, Y.; Matsuzawa, N.; Ogita, H.; Takai, Y. Involvement of the interaction of afadin with ZO-1 in the formation of tight junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 2010, 285, 5003–5012. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Nagafuchi, A.; Moroi, S.; Tsukita, S. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to alpha catenin and actin filaments. J. Cell Biol. 1997, 138, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [PubMed]
- Schneider, M.R.; Kolligs, F.T. E-cadherin’s role in development, tissue homeostasis and disease: Insights from mouse models: Tissue-specific inactivation of the adhesion protein E-cadherin in mice reveals its functions in health and disease. Bioessays 2015, 37, 294–304. [Google Scholar] [CrossRef]
- Bondow, B.J.; Faber, M.L.; Wojta, K.J.; Walker, E.M.; Battle, M.A. E-cadherin is required for intestinal morphogenesis in the mouse. Dev. Biol. 2012, 371, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tinkle, C.L.; Pasolli, H.A.; Stokes, N.; Fuchs, E. New insights into cadherin function in epidermal sheet formation and maintenance of tissue integrity. Proc. Natl. Acad. Sci. USA 2008, 105, 15405–15410. [Google Scholar] [CrossRef] [Green Version]
- Libusova, L.; Stemmler, M.P.; Hierholzer, A.; Schwarz, H.; Kemler, R. N-cadherin can structurally substitute for E-cadherin during intestinal development but leads to polyp formation. Development 2010, 137, 2297–2305. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S. Control of polarized cell morphology and motility by adherens junctions. Semin. Cell Dev. Biol. 2011, 22, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Halbleib, J.M.; Nelson, W.J. Cadherins in development: Cell adhesion, sorting, and tissue morphogenesis. Genes Dev. 2006, 20, 3199–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef]
- Takahashi, K.; Nakanishi, H.; Miyahara, M.; Mandai, K.; Satoh, K.; Satoh, A.; Nishioka, H.; Aoki, J.; Nomoto, A.; Mizoguchi, A.; et al. Nectin/PRR: An immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 1999, 145, 539–549. [Google Scholar] [CrossRef]
- Asakura, T.; Nakanishi, H.; Sakisaka, T.; Takahashi, K.; Mandai, K.; Nishimura, M.; Sasaki, T.; Takai, Y. Similar and differential behaviour between the nectin-afadin-ponsin and cadherin-catenin systems during the formation and disruption of the polarized junctional alignment in epithelial cells. Genes Cells 1999, 4, 573–581. [Google Scholar] [CrossRef]
- Yamada, A.; Fujita, N.; Sato, T.; Okamoto, R.; Ooshio, T.; Hirota, T.; Morimoto, K.; Irie, K.; Takai, Y. Requirement of nectin, but not cadherin, for formation of claudin-based tight junctions in annexin II-knockdown MDCK cells. Oncogene 2006, 25, 5085–5102. [Google Scholar] [CrossRef] [Green Version]
- Takai, Y.; Nakanishi, H. Nectin and afadin: Novel organizers of intercellular junctions. J. Cell Sci. 2003, 116, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Fujita, N.; Yamada, A.; Ooshio, T.; Okamoto, R.; Irie, K.; Takai, Y. Regulation of the assembly and adhesion activity of E-cadherin by nectin and afadin for the formation of adherens junctions in Madin-Darby canine kidney cells. J. Biol. Chem. 2006, 281, 5288–5299. [Google Scholar] [CrossRef] [Green Version]
- Marchiando, A.M.; Graham, W.V.; Turner, J.R. Epithelial barriers in homeostasis and disease. Annu. Rev. Pathol. 2010, 5, 119–144. [Google Scholar] [CrossRef]
- Catalioto, R.M.; Maggi, C.A.; Giuliani, S. Intestinal epithelial barrier dysfunction in disease and possible therapeutical interventions. Curr. Med. Chem. 2011, 18, 398–426. [Google Scholar] [CrossRef] [PubMed]
- Hilsden, R.J.; Meddings, J.B.; Hardin, J.; Gall, D.G.; Sutherland, L.R. Intestinal permeability and postheparin plasma diamine oxidase activity in the prediction of Crohn’s disease relapse. Inflamm. Bowel Dis. 1999, 5, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.; Vogelsang, H.; Hubl, W.; Waldhoer, T.; Lochs, H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 1993, 341, 1437–1439. [Google Scholar] [CrossRef]
- Su, L.; Shen, L.; Clayburgh, D.R.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 2009, 136, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Danese, S.; Fiocchi, C. Ulcerative colitis. N. Engl. J. Med. 2011, 365, 1713–1725. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged >/=18 Years—United States, 2015. Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, G.C.; Chong, C.A.; Chong, R.Y. National estimates of the burden of inflammatory bowel disease among racial and ethnic groups in the United States. J. Crohns Colitis 2014, 8, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Michielan, A.; D’Inca, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediat. Inflamm. 2015, 2015, 628157. [Google Scholar] [CrossRef] [Green Version]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Karayiannakis, A.J.; Syrigos, K.N.; Efstathiou, J.; Valizadeh, A.; Noda, M.; Playford, R.J.; Kmiot, W.; Pignatelli, M. Expression of catenins and E-cadherin during epithelial restitution in inflammatory bowel disease. J. Pathol. 1998, 185, 413–418. [Google Scholar] [CrossRef]
- Bruewer, M.; Samarin, S.; Nusrat, A. Inflammatory bowel disease and the apical junctional complex. Ann. N. Y. Acad. Sci. 2006, 1072, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermuller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Kucharzik, T.; Walsh, S.V.; Chen, J.; Parkos, C.A.; Nusrat, A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am. J. Pathol. 2001, 159, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- Frei, S.M.; Hemsley, C.; Pesch, T.; Lang, S.; Weber, A.; Jehle, E.; Ruhl, A.; Fried, M.; Rogler, G.; Scharl, M. The role for dickkopf-homolog-1 in the pathogenesis of Crohn’s disease-associated fistulae. PLoS ONE 2013, 8, e78882. [Google Scholar] [CrossRef]
- Moparthi, L.; Koch, S. Wnt signaling in intestinal inflammation. Differentiation 2019, 108, 24–32. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Vlad-Fiegen, A.; Langerak, A.; Eberth, S.; Muller, O. The Wnt pathway destabilizes adherens junctions and promotes cell migration via beta-catenin and its target gene cyclin D1. FEBS Open Bio 2012, 2, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Huels, D.J.; Ridgway, R.A.; Radulescu, S.; Leushacke, M.; Campbell, A.D.; Biswas, S.; Leedham, S.; Serra, S.; Chetty, R.; Moreaux, G.; et al. E-cadherin can limit the transforming properties of activating beta-catenin mutations. EMBO J. 2015, 34, 2321–2333. [Google Scholar] [CrossRef]
- Taddei, A.; Giampietro, C.; Conti, A.; Orsenigo, F.; Breviario, F.; Pirazzoli, V.; Potente, M.; Daly, C.; Dimmeler, S.; Dejana, E. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 2008, 10, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Graham, W.V.; Wang, Y.; Witkowski, E.D.; Schwarz, B.T.; Turner, J.R. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 2005, 166, 409–419. [Google Scholar] [CrossRef]
- Al-Sadi, R.M.; Ma, T.Y. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J. Immunol. 2007, 178, 4641–4649. [Google Scholar] [CrossRef] [Green Version]
- Blair, S.A.; Kane, S.V.; Clayburgh, D.R.; Turner, J.R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Investig. J. Tech. Methods Pathol. 2006, 86, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, A.; Berti, I.; Gerarduzzi, T.; Not, T.; Colletti, R.B.; Drago, S.; Elitsur, Y.; Green, P.H.; Guandalini, S.; Hill, I.D.; et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: A large multicenter study. Arch. Intern. Med. 2003, 163, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Junker, Y.; Barisani, D. Celiac disease: From pathogenesis to novel therapies. Gastroenterology 2009, 137, 1912–1933. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Quarsten, H.; Bergseng, E.; Khosla, C.; Sollid, L.M. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc. Natl. Acad. Sci. USA 2004, 101, 4175–4179. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, E.M.; Lundin, K.E.; Krajci, P.; Scott, H.; Sollid, L.M.; Brandtzaeg, P. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut 1995, 37, 766–776. [Google Scholar] [CrossRef] [Green Version]
- Schulzke, J.D.; Bentzel, C.J.; Schulzke, I.; Riecken, E.O.; Fromm, M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatric Res. 1998, 43, 435–441. [Google Scholar] [CrossRef]
- Szakal, D.N.; Gyorffy, H.; Arato, A.; Cseh, A.; Molnar, K.; Papp, M.; Dezsofi, A.; Veres, G. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Arch. 2010, 456, 245–250. [Google Scholar] [CrossRef]
- Schumann, M.; Siegmund, B.; Schulzke, J.D.; Fromm, M. Celiac Disease: Role of the Epithelial Barrier. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Not, T.; Wang, W.; Uzzau, S.; Berti, I.; Tommasini, A.; Goldblum, S.E. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet 2000, 355, 1518–1519. [Google Scholar] [CrossRef]
- Pizzuti, D.; Bortolami, M.; Mazzon, E.; Buda, A.; Guariso, G.; D’Odorico, A.; Chiarelli, S.; D’Inca, R.; De Lazzari, F.; Martines, D. Transcriptional downregulation of tight junction protein ZO-1 in active coeliac disease is reversed after a gluten-free diet. Dig. Liver Dis. 2004, 36, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Finamore, A.; Ara, C.; Di Sabatino, A.; Mengheri, E.; Corazza, G.R. Altered expression, localization, and phosphorylation of epithelial junctional proteins in celiac disease. Am. J. Clin. Pathol. 2006, 125, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Juuti-Uusitalo, K.; Maki, M.; Kainulainen, H.; Isola, J.; Kaukinen, K. Gluten affects epithelial differentiation-associated genes in small intestinal mucosa of coeliac patients. Clin. Exp. Immunol. 2007, 150, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Capuano, M.; Iaffaldano, L.; Tinto, N.; Montanaro, D.; Capobianco, V.; Izzo, V.; Tucci, F.; Troncone, G.; Greco, L.; Sacchetti, L. MicroRNA-449a overexpression, reduced NOTCH1 signals and scarce goblet cells characterize the small intestine of celiac patients. PLoS ONE 2011, 6, e29094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oittinen, M.; Popp, A.; Kurppa, K.; Lindfors, K.; Maki, M.; Kaikkonen, M.U.; Viiri, K. Polycomb Repressive Complex 2 Enacts Wnt Signaling in Intestinal Homeostasis and Contributes to the Instigation of Stemness in Diseases Entailing Epithelial Hyperplasia or Neoplasia. Stem Cells 2017, 35, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Blue, E.K.; Goeckeler, Z.M.; Jin, Y.; Hou, L.; Dixon, S.A.; Herring, B.P.; Wysolmerski, R.B.; Gallagher, P.J. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am. J. Physiol. Cell Physiol. 2002, 282, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 2001, 276, 4527–4530. [Google Scholar] [CrossRef] [Green Version]
- Zhi, G.; Ryder, J.W.; Huang, J.; Ding, P.; Chen, Y.; Zhao, Y.; Kamm, K.E.; Stull, J.T. Myosin light chain kinase and myosin phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. Proc. Natl. Acad. Sci. USA 2005, 102, 17519–17524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, P.J.; Herring, B.P.; Griffin, S.A.; Stull, J.T. Molecular characterization of a mammalian smooth muscle myosin light chain kinase. J. Biol. Chem. 1991, 266, 23936–23944. [Google Scholar] [PubMed]
- Herring, B.P.; El-Mounayri, O.; Gallagher, P.J.; Yin, F.; Zhou, J. Regulation of myosin light chain kinase and telokin expression in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 2006, 291, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Clayburgh, D.R.; Rosen, S.; Witkowski, E.D.; Wang, F.; Blair, S.; Dudek, S.; Garcia, J.G.; Alverdy, J.C.; Turner, J.R. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J. Biol. Chem. 2004, 279, 55506–55513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, K.E.; Turner, J.R. Myosin light chain kinase: Pulling the strings of epithelial tight junction function. Ann. N. Y. Acad. Sci. 2012, 1258, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Zahs, A.; Bird, M.D.; Ramirez, L.; Turner, J.R.; Choudhry, M.A.; Kovacs, E.J. Inhibition of long myosin light-chain kinase activation alleviates intestinal damage after binge ethanol exposure and burn injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Beard, R.S., Jr.; Haines, R.J.; Wu, K.Y.; Reynolds, J.J.; Davis, S.M.; Elliott, J.E.; Malinin, N.L.; Chatterjee, V.; Cha, B.J.; Wu, M.H.; et al. Non-muscle Mlck is required for beta-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1beta-mediated barrier dysfunction in brain endothelial cells. J. Cell Sci. 2014, 127, 1840–1853. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Wang, C.; Shi, L.; Wang, L.; Zhou, Z.; Chen, D.; Wang, J.; Guo, H. Myosin Light Chain Kinase: A Potential Target for Treatment of Inflammatory Diseases. Front. Pharmacol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- He, W.Q.; Zha, J.M.; Wang, Y.T.; Turner, J.R. IgCAM domain 3 is necessary for basal and TNF-induced MLCK1 trafficking in intestinal epithelial cells. FASEB J. 2013, 27, 949. [Google Scholar]
- Graham, W.V.; Wang, F.; Clayburgh, D.R.; Cheng, J.X.; Yoon, B.; Wang, Y.; Lin, A.; Turner, J.R. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J. Biol. Chem. 2006, 281, 26205–26215. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, P.; Su, Q.; Wang, S.; Wang, F. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS ONE 2012, 7, e34946. [Google Scholar] [CrossRef]
- Wu, R.L.; Vazquez-Roque, M.I.; Carlson, P.; Burton, D.; Grover, M.; Camilleri, M.; Turner, J.R. Gluten-induced symptoms in diarrhea-predominant irritable bowel syndrome are associated with increased myosin light chain kinase activity and claudin-15 expression. Lab. Investig. J. Tech. Methods Pathol. 2017, 97, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honjo, M.; Inatani, M.; Kido, N.; Sawamura, T.; Yue, B.Y.; Honda, Y.; Tanihara, H. A myosin light chain kinase inhibitor, ML-9, lowers the intraocular pressure in rabbit eyes. Exp. Eye Res. 2002, 75, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.B.; Cadete, V.J.; Sawicka, J.; Wozniak, M.; Sawicki, G. Effect of the myosin light chain kinase inhibitor ML-7 on the proteome of hearts subjected to ischemia-reperfusion injury. J. Proteom. 2012, 75, 5386–5395. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Kinch, M.S.; Burridge, K. Rho-stimulated contractility contributes to the fibroblastic phenotype of Ras-transformed epithelial cells. Mol. Biol. Cell 1997, 8, 2329–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avizienyte, E.; Fincham, V.J.; Brunton, V.G.; Frame, M.C. Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition. Mol. Biol. Cell 2004, 15, 2794–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, J.H.; Hunter, F.A.; Childs, E.W. PKC and MLCK-dependent, cytokine-induced rat coronary endothelial dysfunction. J. Surg. Res. 2009, 152, 76–83. [Google Scholar] [CrossRef]
- Lukas, T.J.; Mirzoeva, S.; Slomczynska, U.; Watterson, D.M. Identification of novel classes of protein kinase inhibitors using combinatorial peptide chemistry based on functional genomics knowledge. J. Med. Chem. 1999, 42, 910–919. [Google Scholar] [CrossRef]
- Graham, W.V.; He, W.; Marchiando, A.M.; Zha, J.; Singh, G.; Li, H.S.; Biswas, A.; Ong, M.; Jiang, Z.H.; Choi, W.; et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat. Med. 2019, 25, 690–700. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gao, N. RAB and RHO GTPases regulate intestinal crypt cell homeostasis and enterocyte function. Small GTPases 2016, 7, 59–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, A. Rho family GTPases. Biochem. Soc. Trans. 2012, 40, 1378–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J.; Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 1992, 70, 389–399. [Google Scholar] [CrossRef]
- Quiros, M.; Nusrat, A. RhoGTPases, actomyosin signaling and regulation of the epithelial Apical Junctional Complex. Semin. Cell Dev. Biol. 2014, 36, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Besray Unal, E.; Kiel, C.; Benisty, H.; Campbell, A.; Pickering, K.; Bluthgen, N.; Sansom, O.J.; Serrano, L. Systems level expression correlation of Ras GTPase regulators. Cell Commun. Signal. 2018, 16, 46. [Google Scholar] [CrossRef]
- Walsh, S.V.; Hopkins, A.M.; Chen, J.; Narumiya, S.; Parkos, C.A.; Nusrat, A. Rho kinase regulates tight junction function and is necessary for tight junction assembly in polarized intestinal epithelia. Gastroenterology 2001, 121, 566–579. [Google Scholar] [CrossRef]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Morgan-Fisher, M.; Wewer, U.M.; Yoneda, A. Regulation of ROCK activity in cancer. J. Histochem. Cytochem. 2013, 61, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Arnold, T.R.; Stephenson, R.E.; Miller, A.L. Rho GTPases and actomyosin: Partners in regulating epithelial cell-cell junction structure and function. Exp. Cell Res. 2017, 358, 20–30. [Google Scholar] [CrossRef]
- Segain, J.P.; Raingeard de la Bletiere, D.; Sauzeau, V.; Bourreille, A.; Hilaret, G.; Cario-Toumaniantz, C.; Pacaud, P.; Galmiche, J.P.; Loirand, G. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: Evidence in Crohn’s disease and experimental colitis. Gastroenterology 2003, 124, 1180–1187. [Google Scholar] [CrossRef]
- Huang, Y.; Xiao, S.; Jiang, Q. Role of Rho kinase signal pathway in inflammatory bowel disease. Int. J. Clin. Exp. Med. 2015, 8, 3089–3097. [Google Scholar]
- Hofman, P.; Flatau, G.; Selva, E.; Gauthier, M.; Le Negrate, G.; Fiorentini, C.; Rossi, B.; Boquet, P. Escherichia coli cytotoxic necrotizing factor 1 effaces microvilli and decreases transmigration of polymorphonuclear leukocytes in intestinal T84 epithelial cell monolayers. Infect. Immun. 1998, 66, 2494–2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiaolu, D.; Jing, P.; Fang, H.; Lifen, Y.; Liwen, W.; Ciliu, Z.; Fei, Y. Role of p115RhoGEF in lipopolysaccharide-induced mouse brain microvascular endothelial barrier dysfunction. Brain Res. 2011, 1387, 1–7. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Peng, J.; Deng, X.L.; Yang, L.F.; Wu, L.W.; Zhang, C.L.; Yin, F. RhoA and NF-kappaB are involved in lipopolysaccharide-induced brain microvascular cell line hyperpermeability. Neuroscience 2011, 188, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Bellovin, D.I.; Bates, R.C.; Muzikansky, A.; Rimm, D.L.; Mercurio, A.M. Altered localization of p120 catenin during epithelial to mesenchymal transition of colon carcinoma is prognostic for aggressive disease. Cancer Res. 2005, 65, 10938–10945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohn, M.R.; Brown, M.V.; Reynolds, A.B. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 2009, 184, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, M.; Huveldt, D.; Kreinest, P.; Lohse, C.M.; Cheville, J.C.; Parker, A.S.; Copland, J.A.; Anastasiadis, P.Z. A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J. Biol. Chem. 2008, 283, 18344–18354. [Google Scholar] [CrossRef] [Green Version]
- de Toledo, M.; Anguille, C.; Roger, L.; Roux, P.; Gadea, G. Cooperative anti-invasive effect of Cdc42/Rac1 activation and ROCK inhibition in SW620 colorectal cancer cells with elevated blebbing activity. PLoS ONE 2012, 7, e48344. [Google Scholar] [CrossRef] [Green Version]
- Sakamori, R.; Yu, S.; Zhang, X.; Hoffman, A.; Sun, J.; Das, S.; Vedula, P.; Li, G.; Fu, J.; Walker, F.; et al. CDC42 inhibition suppresses progression of incipient intestinal tumors. Cancer Res. 2014, 74, 5480–5492. [Google Scholar] [CrossRef] [Green Version]
- Asano, T.; Ikegaki, I.; Satoh, S.; Suzuki, Y.; Shibuya, M.; Takayasu, M.; Hidaka, H. Mechanism of action of a novel antivasospasm drug, HA1077. J. Pharmacol. Exp. Ther. 1987, 241, 1033–1040. [Google Scholar]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997, 389, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Yu, J.; Gao, Y.; Kumar, G.; Guo, M.; Zhao, Y.; Fang, Q.; Zhang, H.; Yu, J.; Jiang, Y.; et al. Therapeutic potentials of the Rho kinase inhibitor Fasudil in experimental autoimmune encephalomyelitis and the related mechanisms. Metab. Brain Dis. 2019, 34, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Palecek, J.; Zweigerdt, R.; Olmer, R.; Martin, U.; Kirschning, A.; Drager, G. A practical synthesis of Rho-Kinase inhibitor Y-27632 and fluoro derivatives and their evaluation in human pluripotent stem cells. Org. Biomol. Chem. 2011, 9, 5503–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rikitake, Y.; Kim, H.H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Zheng, Y.; von Bornstadt, D.; Wei, Y.; Balcioglu, A.; Daneshmand, A.; Yalcin, N.; Yu, E.; Herisson, F.; Atalay, Y.B.; et al. Selective ROCK2 Inhibition In Focal Cerebral Ischemia. Ann. Clin. Transl. Neurol. 2014, 1, 2–14. [Google Scholar]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Du, L.; Kim, J.J.; Shen, J.; Dai, N. Crosstalk between Inflammation and ROCK/MLCK Signaling Pathways in Gastrointestinal Disorders with Intestinal Hyperpermeability. Gastroenterol Res. Pract. 2016, 2016, 7374197. [Google Scholar] [CrossRef] [Green Version]
- Zolotarevsky, Y.; Hecht, G.; Koutsouris, A.; Gonzalez, D.E.; Quan, C.; Tom, J.; Mrsny, R.J.; Turner, J.R. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002, 123, 163–172. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Blikslager, A.T. The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases. Int. J. Mol. Sci. 2020, 21, 3550. https://doi.org/10.3390/ijms21103550
Jin Y, Blikslager AT. The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases. International Journal of Molecular Sciences. 2020; 21(10):3550. https://doi.org/10.3390/ijms21103550
Chicago/Turabian StyleJin, Younggeon, and Anthony T. Blikslager. 2020. "The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases" International Journal of Molecular Sciences 21, no. 10: 3550. https://doi.org/10.3390/ijms21103550
APA StyleJin, Y., & Blikslager, A. T. (2020). The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases. International Journal of Molecular Sciences, 21(10), 3550. https://doi.org/10.3390/ijms21103550