Intronic Alternative Polyadenylation in the Middle of the DMD Gene Produces Half-Size N-Terminal Dystrophin with a Potential Implication of ECG Abnormalities of DMD Patients

 , , , and
, , , and
Abstract
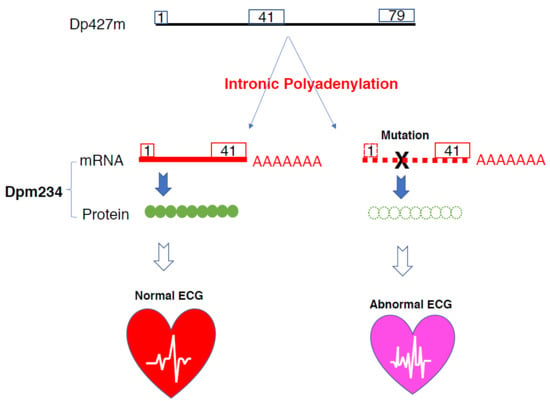
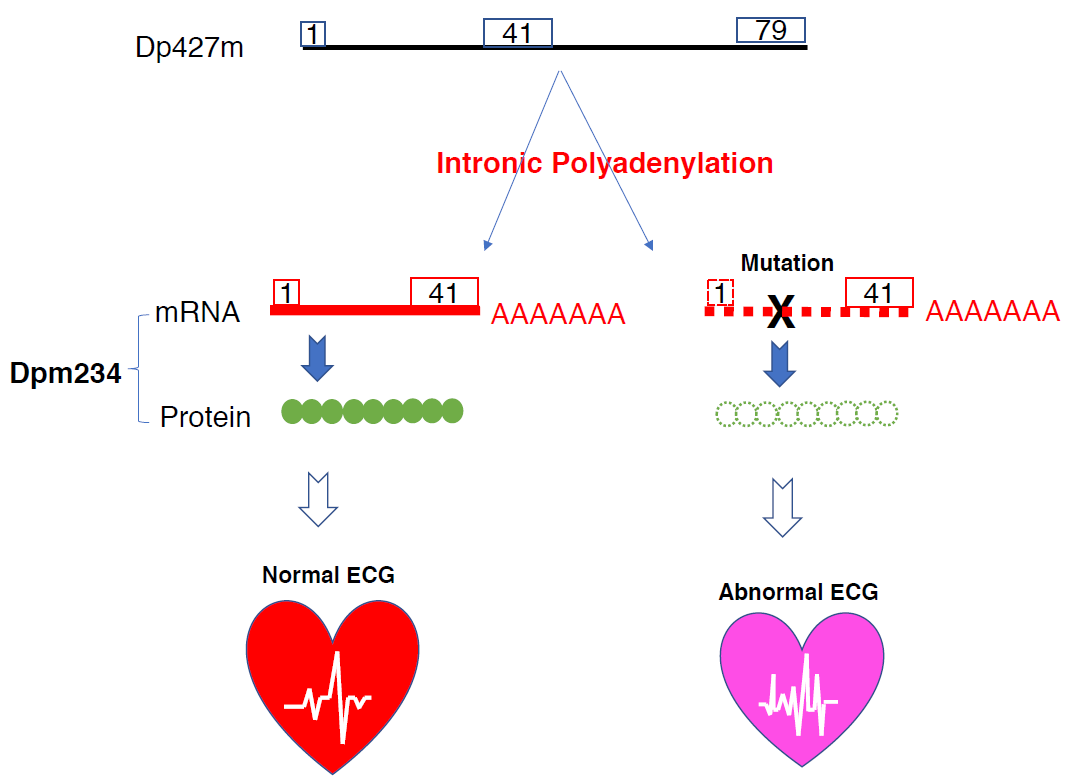
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
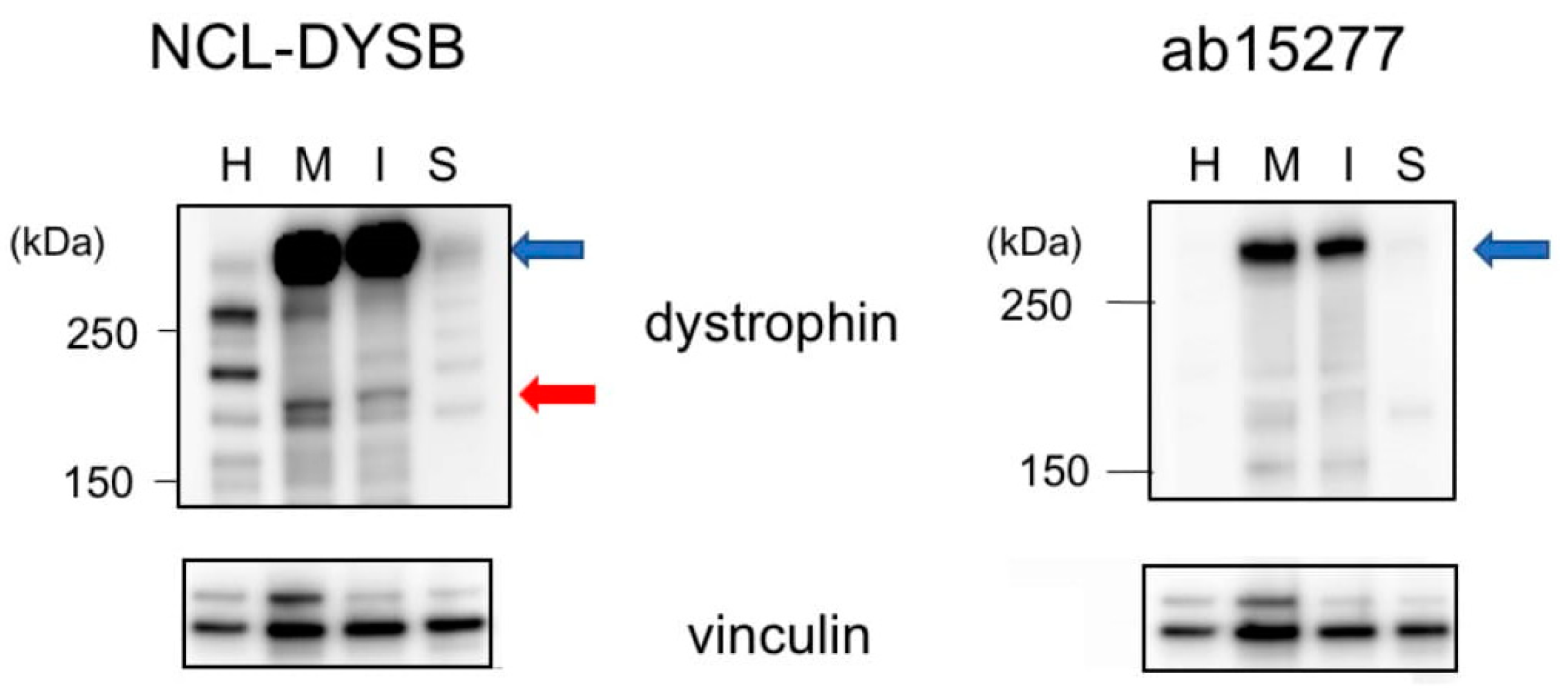{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Studies
2.1.1. Cell Lines
2.1.2. cDNA Synthesis from Total RNA
2.1.3. Cloning of a Novel Transcript by 3′-RACE
2.1.4. Protein Sample Preparation
2.1.5. Protein Analysis
2.2. Clinical Studies
2.2.1. Patients
2.2.2. ECG Examination
2.2.3. Echocardiography
2.2.4. Statistics
2.2.5. Ethics
3. Results
3.1. Cloning of a Novel 3′ end Exon of DMD Transcript by 3′RACE
3.2. Identification of the Novel DMD Transcript in Cardiomyocytes
3.3. Protein Products of the Cloned Transcripts
3.4. Identification of the Dpm234 Protein
3.5. Cardiac Findings in DMD Patients
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bulman, D.E.; Murphy, E.G.; Zubrzycka-Gaarn, E.E.; Worton, R.G.; Ray, P.N. Differentiation of Duchenne and Becker muscular dystrophy phenotypes with amino- and carboxy-terminal antisera specific for dystrophin. Am. J. Hum. Genet. 1991, 48, 295–304. [Google Scholar] [PubMed]
- Helliwell, T.R.; Ellis, J.M.; Mountford, R.C.; Appleton, R.E.; Morris, G.E. A truncated dystrophin lacking the C-terminal domains is localized at the muscle membrane. Am. J. Hum. Genet. 1992, 50, 508–514. [Google Scholar] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Massourides, E.; Polentes, J.; Mangeot, P.E.; Mournetas, V.; Nectoux, J.; Deburgrave, N.; Nusbaum, P.; Leturcq, F.; Popplewell, L.; Dickson, G.; et al. Dp412e: A novel human embryonic dystrophin isoform induced by BMP4 in early differentiated cells. Skelet Muscle 2015, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feener, C.A.; Koenig, M.; Kunkel, L.M. Alternative splicing of human dystrophin mRNA generates isoforms at the carboxy terminus. Nature 1989, 338, 509–511. [Google Scholar] [CrossRef]
- Rau, F.; Laine, J.; Ramanoudjame, L.; Ferry, A.; Arandel, L.; Delalande, O.; Jollet, A.; Dingli, F.; Lee, K.Y.; Peccate, C.; et al. Abnormal splicing switch of DMD’s penultimate exon compromises muscle fibre maintenance in myotonic dystrophy. Nat. Commun. 2015, 6, 7205. [Google Scholar] [CrossRef] [Green Version]
- Nishida, A.; Minegishi, M.; Takeuchi, A.; Awano, H.; Niba, E.T.; Matsuo, M. Neuronal SH-SY5Y cells use the C-dystrophin promoter coupled with exon 78 skipping and display multiple patterns of alternative splicing including two intronic insertion events. Hum. Genet. 2015, 134, 993–1001. [Google Scholar] [CrossRef]
- Rani, A.Q.M.; Farea, M.; Maeta, K.; Kawaguchi, T.; Awano, H.; Nagai, M.; Nishio, H.; Matsuo, M. Identification of the shortest splice variant of Dp71, together with five known variants, in glioblastoma cells. Biochem. Biophy. Res. Comm. 2019, 508, 640–645. [Google Scholar] [CrossRef]
- Rani, A.; Maeta, K.; Kawaguchi, T.; Awano, H.; Nagai, M.; Nishio, H.; Matsuo, M. Schwann cell-specific Dp116 is expressed in glioblastoma cells, revealing two novel DMD gene splicing patterns. Biochem. Biophys. Rep. 2019, 20, 100703. [Google Scholar]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Tinsley, J.; Blake, D.; davies, K. Apo-dystrophin-3: A 2.2 kb transcript from the DMD locusencoding the dystrophin glycoprotein binding site. Hum. Mol. Genet. 1993, 2, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Miura, T.; Aoyagi, T.; Ogata, H.; Hamada, S.; Minami, R. Duchenne muscular dystrophy: Survival by cardio-respiratory interventions. Neuromuscul. Disord. 2011, 21, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Ararat, E.; Mhanna, M.J. Cardiac phenotype determines survival in Duchenne muscular dystrophy. Pediatr. Pulmonol. 2016, 51, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Eidem, B.W.; Belmont, J.W.; Craigen, W.J.; Ware, S.M.; Fernbach, S.D.; Neish, S.R.; Smith, E.O.; Towbin, J.A. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005, 112, 2799–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwath, M.L.; Jacobs, I.B.; Crowe, C.A.; Ashwath, R.C.; Super, D.M.; Bahler, R.C. Left Ventricular Dysfunction in Duchenne Muscular Dystrophy and Genotype. Am. J. Cardiol. 2014, 114, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, M.; Awano, H.; Matsumoto, M.; Nagai, M.; Kawaguchi, T.; Zhang, Z.; Nishio, H. Dystrophin Dp116: A yet to Be Investigated Product of the Duchenne Muscular Dystrophy Gene. Genes 2017, 8, 251. [Google Scholar] [CrossRef]
- Yamamoto, T.; Awano, H.; Zhan, Z.; Enomoto-Sakuma, M.; Kitaaki, S.; Matsumoto, M.; Nagai, M.; Sato, I.; Imanishi, T.; Hayashi, N.; et al. Cardiac dysfunction in Duchenne muscular dystrophy is less frequent in patients with mutations in the dystrophin Dp116 coding region than in other regions. Circ. Genome Precis. Med. 2018, 11, e001782. [Google Scholar] [CrossRef] [Green Version]
- Weisenfeld, S.; Messinger, W. Cardiac involvement in progressive muscular dystrophy. Am. Heart J. 1952, 43, 170–187. [Google Scholar] [CrossRef]
- Heymsfield, S.B.; McNish, T.; Perkins, J.V.; Felner, J.M. Sequence of cardiac changes in Duchenne muscular dystrophy. Am. Heart J. 1978, 95, 283–294. [Google Scholar] [CrossRef]
- Takami, Y.; Takeshima, Y.; Awano, H.; Okizuka, Y.; Yagi, M.; Matsuo, M. High incidence of electrocardiogram abnormalities in young patients with duchenne muscular dystrophy. Pediatr. Neurol. 2008, 39, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Fitch, C.W.; Ainger, L.E. The Frank vectorcardiogram and the electrocardiogram in Duchenne progressive muscular dystrophy. Circulation 1967, 35, 1124–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, S.K.; Johnson, W.W. Cardiac conduction abnormalities in children with Duchenne’s progressive muscular dystrophy: Electrocardiographic features and morphologic correlates. Circulation 1982, 66, 853–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.; Kinnett, K.; Wang, Y.; Ittenbach, R.F.; Benson, D.W.; Cripe, L. Electrocardiographic abnormalities in very young Duchenne muscular dystrophy patients precede the onset of cardiac dysfunction. Neuromuscul. Disord. 2011, 21, 462–467. [Google Scholar] [CrossRef]
- Matsuo, M.; Masumura, T.; Nishio, H.; Nakajima, T.; Kitoh, Y.; Takumi, T.; Koga, J.; Nakamura, H. Exon skipping during splicing of dystrophin mRNA precursor due to an intraexon deletion in the dystrophin gene of Duchenne muscular dystrophy kobe. J. Clin. Investig. 1991, 87, 2127–2131. [Google Scholar] [CrossRef]
- Tran, V.K.; Zhang, Z.; Yagi, M.; Nishiyama, A.; Habara, Y.; Takeshima, Y.; Matsuo, M. A novel cryptic exon identified in the 3′ region of intron 2 of the human dystrophin gene. J. Hum. Genet. 2005, 50, 425–433. [Google Scholar] [CrossRef]
- Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef] [Green Version]
- Molina, K.M.; Shrader, P.; Colan, S.D.; Mital, S.; Margossian, R.; Sleeper, L.A.; Shirali, G.; Barker, P.; Canter, C.E.; Altmann, K.; et al. Predictors of disease progression in pediatric dilated cardiomyopathy. Circ. Heart Fail. 2013, 6, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Kodippili, K.; Yue, Y.; Hakim, C.H.; Wasala, L.; Pan, X.; Zhang, K.; Yang, N.N.; Duan, D.; Lai, Y. Dystrophin contains multiple independent membrane-binding domains. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.; t Hoen, P.A.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorenweerd, N.; Mahfouz, A.; van Putten, M.; Kaliyaperumal, R.; T’ Hoen, P.A.C.; Hendriksen, J.G.M.; Aartsma Rus, A.; Verschuuren, J.J.G.M.; Niks, E.; Reinders, M.J.T.; et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 12575. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zheng, D.; Wei, L.; Ding, Q.; Tian, B. Regulation of Intronic Polyadenylation by PCF11 Impacts mRNA Expression of Long Genes. Cell Rep. 2019, 26, 2766–2778.e2766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoekstra, M.; Mummery, C.; Wilde, A.A.M.; Bezzina, C.; Verkerk, A. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front. Physiol. 2012, 3, 346. [Google Scholar] [CrossRef] [Green Version]
- Pillers, D.A.; Bulman, D.E.; Welber, R.G.; Sigesmund, D.A.; Musarella, M.A.; Powell, B.R.; Murphey, W.H.; Westall, C.; Panton, C.; Becker, L.E.; et al. Dystrophin expression in the human retina is required for normal function as defined by electroretinography. Nat. Genet 1993, 4, 82–86. [Google Scholar] [CrossRef]
- Ricotti, V.; Jagle, H.; Theodorou, M.; Moore, A.T.; Muntoni, F.; Thompson, D.A. Ocular and neurodevelopmental features of Duchenne muscular dystrophy: A signature of dystrophin function in the central nervous system. Eur. J. Hum. Genet. 2016, 24, 562–568. [Google Scholar] [CrossRef]
- Moizard, M.P.; Toutain, A.; Fournier, D.; Berret, F.; Raynaud, M.; Billard, C.; Andres, C.; Moraine, C. Severe cognitive impairment in DMD: Obvious clinical indication for Dp71 isoform point mutation screening. Eur. J. Hum. Genet. 2000, 8, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Claudepierre, T.; Mornet, D.; Pannicke, T.; Forster, V.; Dalloz, C.; Bolanos, F.; Sahel, J.; Reichenbach, A.; Rendon, A. Expression of Dp71 in Muller glial cells: A comparison with utrophin- and dystrophin-associated proteins. Investig. Ophthalmol. Vis. Sci. 2000, 41, 294–304. [Google Scholar]
- Matsumoto, M.; Awano, H.; Lee, T.; Takeshima, Y.; Matsuo, M.; Iijima, K. Patients with Duchenne muscular dystrophy are significantly shorter than those with Becker muscular dystrophy, with the higher incidence of short stature in Dp71 mutated subgroup. Neuromuscul. Disord. 2017, 27, 1023–1028. [Google Scholar] [CrossRef]
- Eisen, B.; Ben Jehuda, R.; Cuttitta, A.; Mekies, L.; Shemer, Y.; Baskin, P.; Reiter, I.; Willi, L.; Freimark, D.; Gherghiceanu, M.; et al. Electrophysiological abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from Duchenne muscular dystrophy patients. J. Cell. Mol. Med. 2019, 23, 2125–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Ari, M.; Schick, R.; Barad, L.; Novak, A.; Ben Ari, E.; Lorber, A.; Itskovitz Eldor, J.; Rosen, M.; Weissman, A.; Binah, O. From beat rate variability in induced pluripotent stem cell-derived pacemaker cells to heart rate variability in human subjects. Heart Rhythm 2014, 11, 1808–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, M.; Johnson, E.L.; Coller, H.A. Alternative polyadenylation can regulate post-translational membrane localization. Trends Cell. Mol. Biol. 2015, 10, 37–47. [Google Scholar] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rani, A.Q.M.; Yamamoto, T.; Kawaguchi, T.; Maeta, K.; Awano, H.; Nishio, H.; Matsuo, M. Intronic Alternative Polyadenylation in the Middle of the DMD Gene Produces Half-Size N-Terminal Dystrophin with a Potential Implication of ECG Abnormalities of DMD Patients. Int. J. Mol. Sci. 2020, 21, 3555. https://doi.org/10.3390/ijms21103555
Rani AQM, Yamamoto T, Kawaguchi T, Maeta K, Awano H, Nishio H, Matsuo M. Intronic Alternative Polyadenylation in the Middle of the DMD Gene Produces Half-Size N-Terminal Dystrophin with a Potential Implication of ECG Abnormalities of DMD Patients. International Journal of Molecular Sciences. 2020; 21(10):3555. https://doi.org/10.3390/ijms21103555
Chicago/Turabian StyleRani, Abdul Qawee Mahyoob, Tetsushi Yamamoto, Tatsuya Kawaguchi, Kazuhiro Maeta, Hiroyuki Awano, Hisahide Nishio, and Masafumi Matsuo. 2020. "Intronic Alternative Polyadenylation in the Middle of the DMD Gene Produces Half-Size N-Terminal Dystrophin with a Potential Implication of ECG Abnormalities of DMD Patients" International Journal of Molecular Sciences 21, no. 10: 3555. https://doi.org/10.3390/ijms21103555
APA StyleRani, A. Q. M., Yamamoto, T., Kawaguchi, T., Maeta, K., Awano, H., Nishio, H., & Matsuo, M. (2020). Intronic Alternative Polyadenylation in the Middle of the DMD Gene Produces Half-Size N-Terminal Dystrophin with a Potential Implication of ECG Abnormalities of DMD Patients. International Journal of Molecular Sciences, 21(10), 3555. https://doi.org/10.3390/ijms21103555