Examining Short-Term Responses to a Long-Term Problem: RNA-Seq Analyses of Iron Deficiency Chlorosis Tolerant Soybean

 ,
,
Abstract
:1. Introduction
2. Results
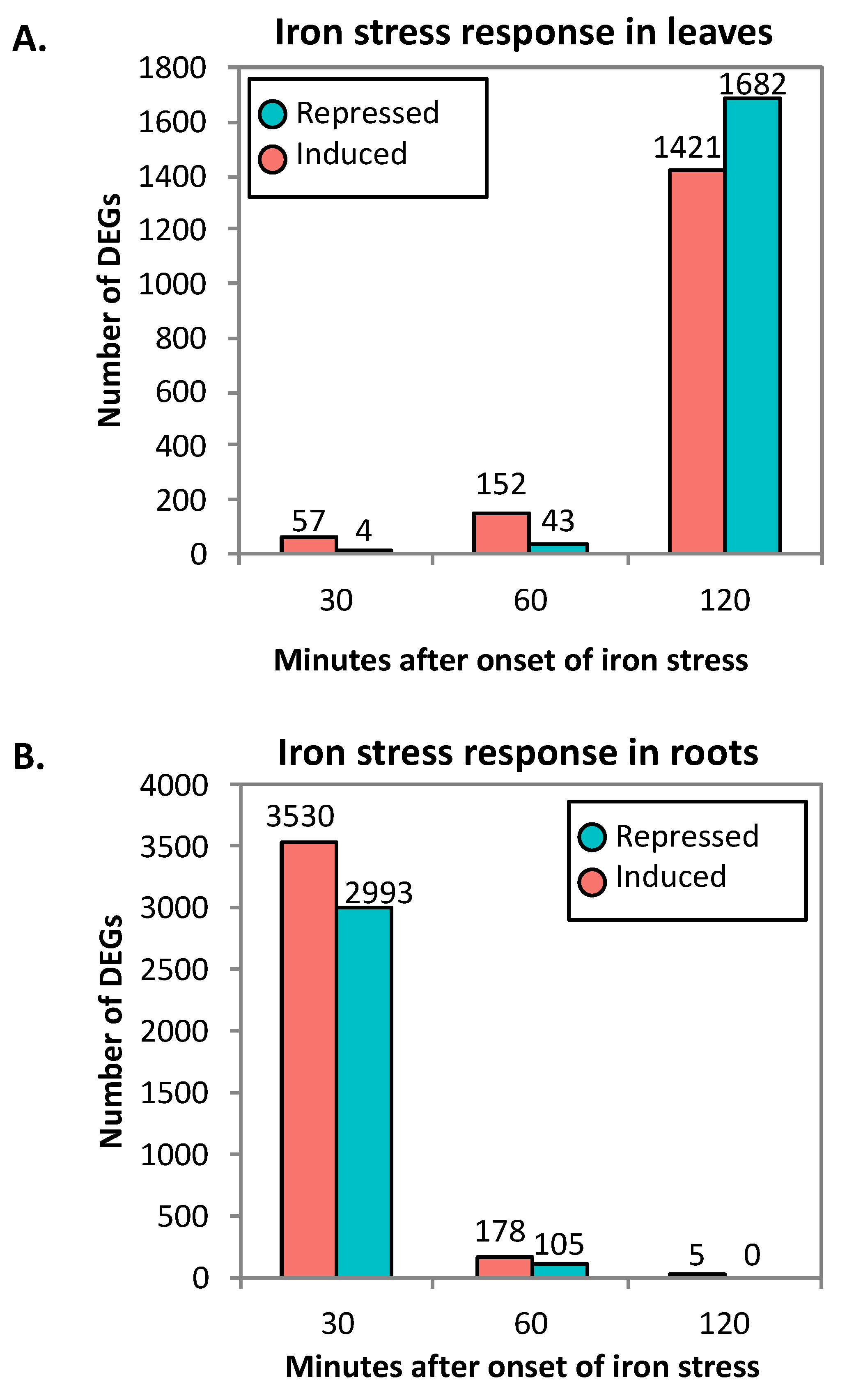
2.1. RNA-Seq Analyses
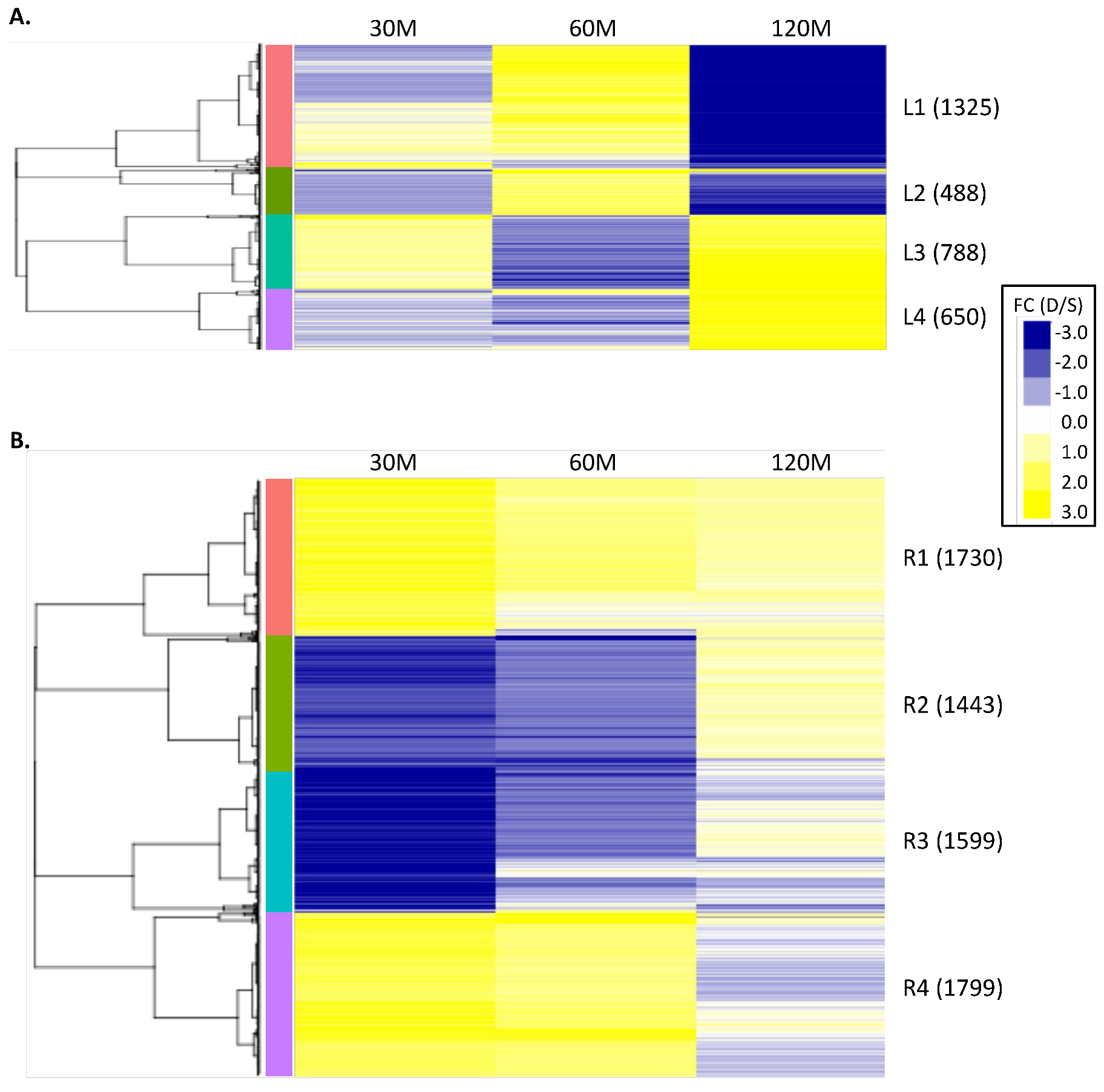
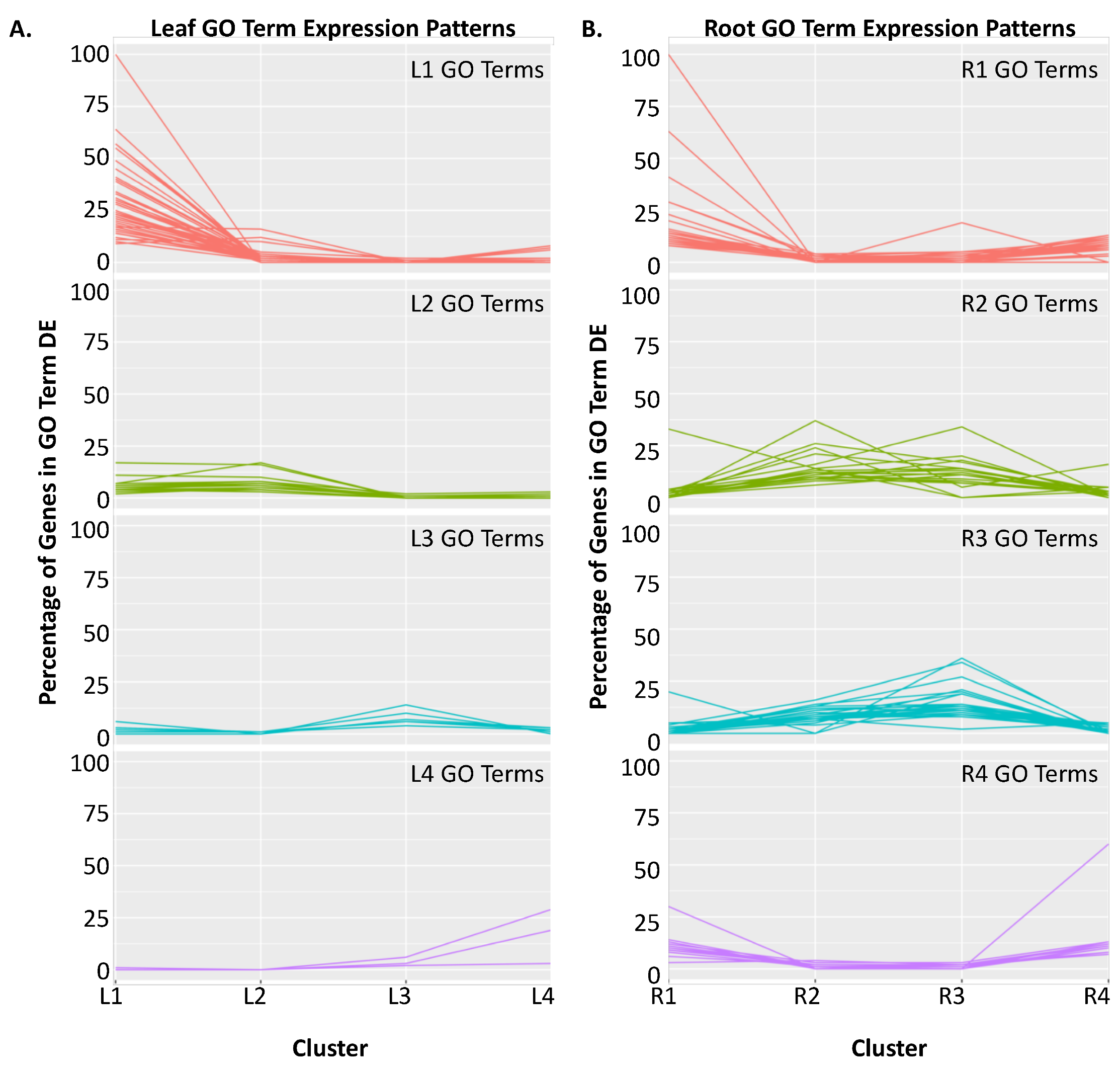
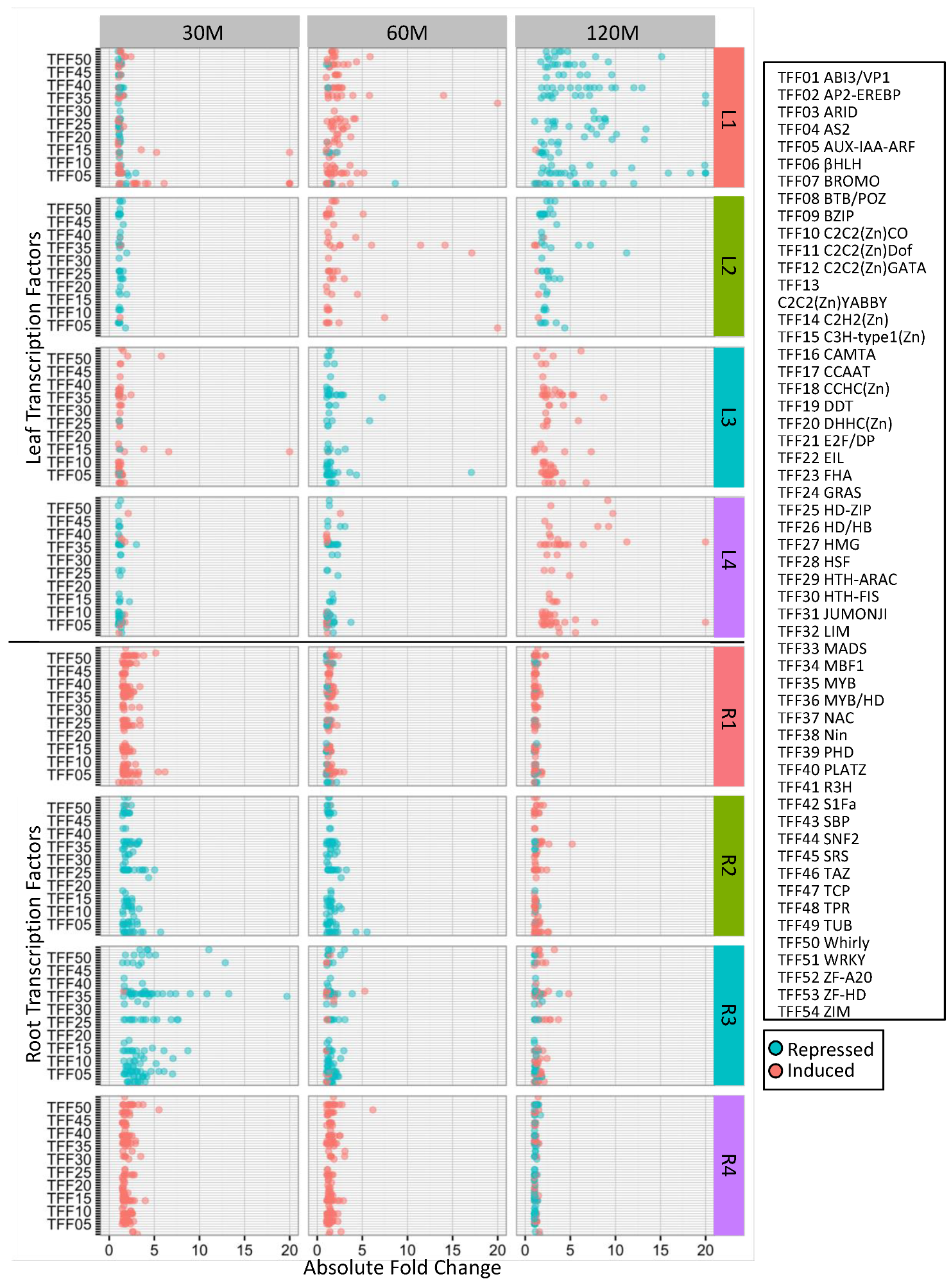
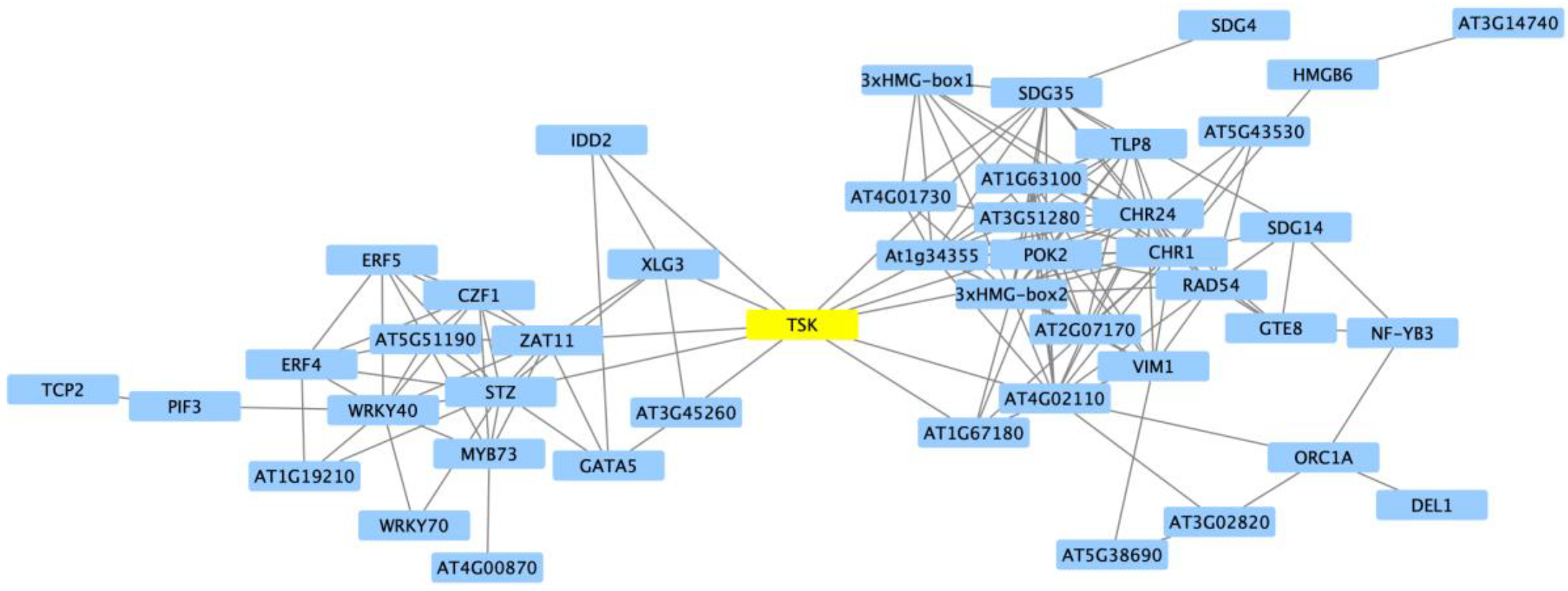
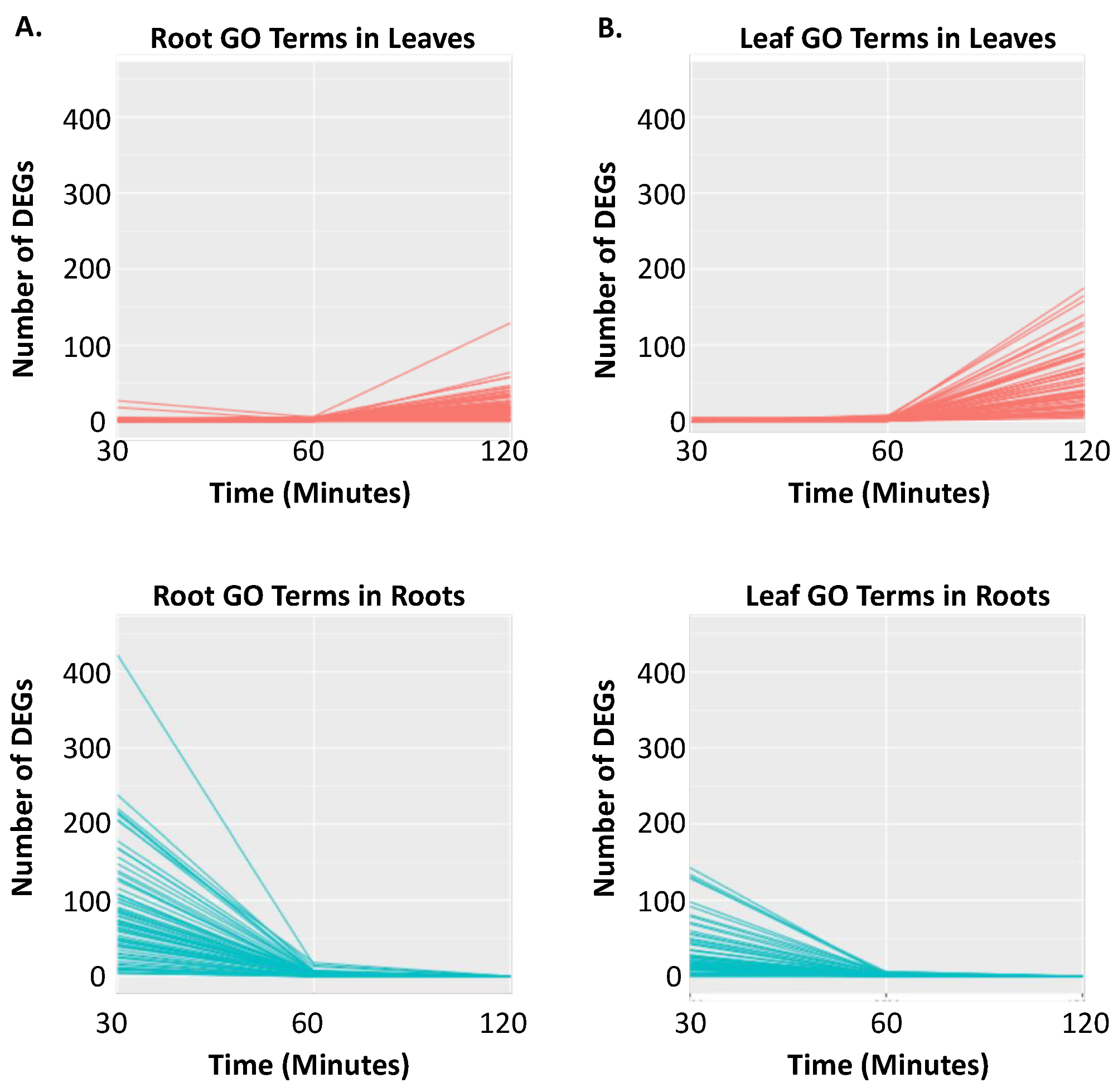
2.2. Clustering of DEGs Based on Expression Pattern Links Gene Function, Expression and Regulation during Iron Stress
3. Discussion
4. Materials and Methods
4.1. Growth Conditions
4.2. RNA Isolation
4.3. RNA-Seq and Data Analysis
4.4. Gene Annotation
4.5. Hierarchical Clustering and Heat Maps
4.6. Identification of Overrepresented Gene Ontology Terms and Transcription Factor Families
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFC | Average Fold Change |
| DEGs | Differentially expressed genes |
| FC | Fold Change |
| FDR | False discovery rate |
| GO | Gene ontology |
| IDC | Iron deficiency chlorosis |
| P | Probability |
| TF | Transcription factor |
| TFFs | Transcription factor families |
| TMM | Trimmed mean of M-values |
References
- Wollman, F.A.; Minai, L.; Nechushtai, R. The biogenesis and assembly of photosynthetic proteins in thylakoid membranes. Biochim. Biophys. Acta 1999, 1411, 21–85. [Google Scholar] [CrossRef] [Green Version]
- Shikanai, T.; Muller-Moule, P.; Munekage, Y.; Niyogi, K.K.; Pilon, M. PAA1, a P-type ATPase of Arabidopsis, functions in copper transport in chloroplasts. Plant Cell 2003, 15, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Hansen, N.C.; Jolley, V.D.; Naeve, S.L.; Goos, R.J. Iron deficiency of soybean in the north central US and associated soil properties. Soil Sci. Plant Nutr. 2004, 50, 983–987. [Google Scholar] [CrossRef]
- Goos, J.R.; Johnson, B. Seed treatment, seeding rate and cultivar effects on iron deficiency chlorosis of soybean. J. Plant Nutr. 2001, 24, 1255–1268. [Google Scholar] [CrossRef]
- Vert, G.A.; Briat, J.-F.; Curie, C. Dual regulation of the Arabidopsis high-affinity root iron uptake system by local and long-distance signals. Plant Physiol. 2003, 132, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, Y.; Goto, F. Long-distance signaling of iron deficiency in plants. Plant Signal. Behav. 2008, 3, 396–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giehl, R.F.; Lima, J.E.; von Wirén, N. Localized iron supply triggers lateral root elongation in Arabidopsis by altering the AUX1-mediated auxin distribution. Plant Cell 2012, 24, 33–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Cózatl, D.G.; Xie, Q.; Akmakjian, G.Z.; Jobe, T.O.; Patel, A.; Stacey, M.G.; Song, L.; Demoin, D.W.; Jurisson, S.S.; Stacey, G. OPT3 is a component of the iron-signaling network between leaves and roots and misregulation of OPT3 leads to an over-accumulation of cadmium in seeds. Mol. Plant 2014, 7, 1455–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Z.; Gayomba, S.R.; Jung, H.-i.; Vimalakumari, N.K.; Piñeros, M.; Craft, E.; Rutzke, M.A.; Danku, J.; Lahner, B.; Punshon, T. OPT3 is a phloem-specific iron transporter that is essential for systemic iron signaling and redistribution of iron and cadmium in Arabidopsis. Plant Cell 2014, 26, 2249–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirayama, T.; Lei, G.J.; Yamaji, N.; Nakagawa, N.; Ma, J.F. The putative peptide gene FEP1 regulates iron deficiency response in Arabidopsis. Plant Cell Physiol. 2018, 59, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, E.P.; Guerinot, M.L. The essential basic helix-loop-helix protein FIT1 is required for the iron deficiency response. Plant Cell 2004, 16, 3400–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.J.; Procter, C.M.; Connolly, E.L.; Guerinot, M.L. A ferric-chelate reductase for iron uptake from soils. Nature 1999, 397, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Vert, G.; Grotz, N.; Dédaldéchamp, F.; Gaymard, F.; Guerinot, M.L.; Briat, J.-F.; Curie, C. IRT1, an Arabidopsis transporter essential for iron uptake from the soil and for plant growth. Plant Cell 2002, 14, 1223–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, N.B.; Giehl, R.F.; Döll, S.; Mock, H.-P.; Strehmel, N.; Scheel, D.; Kong, X.; Hider, R.C.; von Wirén, N. Feruloyl-CoA 6′-Hydroxylase1-dependent coumarins mediate iron acquisition from alkaline substrates in Arabidopsis. Plant Physiol. 2014, 164, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Briat, J.-F.; Curie, C.; Gaymard, F. Iron utilization and metabolism in plants. Curr. Opin. Plant Biol. 2007, 10, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Grotz, N.; Guerinot, M.L. Molecular aspects of Cu, Fe and Zn homeostasis in plants. Biochim. Biophys. Acta 2006, 1763, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Stephan, U.W.; Scholz, G. Nicotianamine: Mediator of transport of iron and heavy metals in the phloem? Physiol. Plant 1993, 88, 522–529. [Google Scholar] [CrossRef]
- García, M.J.; Suárez, V.; Romera, F.J.; Alcántara, E.; Pérez-Vicente, R. A new model involving ethylene, nitric oxide and Fe to explain the regulation of Fe-acquisition genes in Strategy I plants. Plant Physiol. Biochem. 2011, 49, 537–544. [Google Scholar] [CrossRef]
- Liu, X.X.; He, X.L.; Jin, C.W. Roles of chemical signals in regulation of the adaptive responses to iron deficiency. Plant Signal. Behav. 2016, 11, e1179418. [Google Scholar] [CrossRef] [Green Version]
- Wild, M.; Davière, J.-M.; Regnault, T.; Sakvarelidze-Achard, L.; Carrera, E.; Diaz, I.L.; Cayrel, A.; Dubeaux, G.; Vert, G.; Achard, P. Tissue-specific regulation of gibberellin signaling fine-tunes Arabidopsis iron-deficiency responses. Dev. Cell 2016, 37, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Yang, Y.; Liu, K.; Zhang, L.; Guo, H.; Sun, T.; Wang, H. Involvement of endogenous salicylic acid in iron-deficiency responses in Arabidopsis. J. Exp. Bot. 2016, 67, 4179–4193. [Google Scholar] [CrossRef] [Green Version]
- Gayomba, S.R.; Zhai, Z.; Jung, H.-I.; Vatamaniuk, O.K. Local and systemic signaling of iron status and its interactions with homeostasis of other essential elements. Front. Plant Sci. 2015, 6, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.E.; O’Rourke, J.A.; Peiffer, G.A.; Yin, T.; Majumder, M.; Zhang, C.; Cianzio, S.R.; Hill, J.H.; Cook, D.; Whitham, S.A.; et al. Replication protein A subunit 3 and the iron efficiency response in soybean. Plant Cell Environ. 2014, 37, 213–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckhout, T.J.; Yang, T.J.; Schmidt, W. Early iron-deficiency-induced transcriptional changes in Arabidopsis roots as revealed by microarray analyses. BMC Genom. 2009, 10, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran Lauter, A.N.; Peiffer, G.A.; Yin, T.; Whitham, S.A.; Cook, D.; Shoemaker, R.C.; Graham, M.A. Identification of candidate genes involved in early iron deficiency chlorosis signaling in soybean (Glycine max) roots and leaves. BMC Genom. 2014, 15, 702. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.A.; Charlson, D.V.; Gonzalez, D.O.; Vodkin, L.O.; Graham, M.A.; Cianzio, S.R.; Grusak, M.A.; Shoemaker, R.C. Microarray analysis of iron deficiency chlorosis in near-isogenic soybean lines. BMC Genom. 2007, 8, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, J.A.; Nelson, R.T.; Grant, D.; Schmutz, J.; Grimwood, J.; Cannon, S.; Vance, C.P.; Graham, M.A.; Shoemaker, R.C. Integrating microarray analysis and the soybean genome to understand the soybeans iron deficiency response. BMC Genom. 2009, 10, 376. [Google Scholar] [CrossRef] [Green Version]
- Peiffer, G.A.; King, K.E.; Severin, A.J.; May, G.D.; Cianzio, S.R.; Lin, S.F.; Lauter, N.C.; Shoemaker, R.C. Identification of candidate genes underlying an iron efficiency quantitative trait locus in soybean. Plant Physiol. 2012, 158, 1745–1754. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.J.; Waters, B.M. Use of natural variation reveals core genes in the transcriptome of iron-deficient Arabidopsis thaliana roots. J. Exp. Bot. 2012, 63, 1039–1055. [Google Scholar] [CrossRef] [Green Version]
- Waters, B.M.; McInturf, S.A.; Amundsen, K. Transcriptomic and physiological characterization of the fefe mutant of melon (Cucumis melo) reveals new aspects of iron-copper crosstalk. New Phytol. 2014, 203, 1128–1145. [Google Scholar] [CrossRef] [Green Version]
- Waters, B.M.; McInturf, S.A.; Stein, R.J. Rosette iron deficiency transcript and microRNA profiling reveals links between copper and iron homeostasis in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 5903–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.A. The Design of Experiments, 8th ed.; London Oliver & Boyd: Edinburgh, UK, 1966; p. vx. 248p. [Google Scholar]
- Bonferroni, C.E. Ill Calcolo delle assicurazioni su gruppi di teste. In Studi in Onore del Professore Salvatore Ortu Carboni; Bardi: Rome, Italy, 1935; pp. 13–60. [Google Scholar]
- Berardini, T.Z.; Mundodi, S.; Reiser, L.; Huala, E.; Garcia-Hernandez, M.; Zhang, P.; Mueller, L.A.; Yoon, J.; Doyle, A.; Lander, G.; et al. Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol. 2004, 135, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Libault, M.; Joshi, T.; Valliyodan, B.; Nguyen, H.T.; Xu, D.; Stacey, G.; Cheng, J. SoyDB: A knowledge database of soybean transcription factors. BMC Plant Biol. 2010, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Castro-Guerrero, N.A.; McInturf, S.A.; Nguyen, N.T.; Dame, A.N.; Wang, J.; Bindbeutel, R.K.; Joshi, T.; Jurisson, S.S.; Nusinow, D.A.; et al. Changes in iron availability in Arabidopsis are rapidly sensed in the leaf vasculature and impaired sensing leads to opposite transcriptional programs in leaves and roots. Plant Cell Environ. 2018, 41, 2263–2276. [Google Scholar] [CrossRef]
- Rodríguez-Celma, J.; Lin, W.-D.; Fu, G.-M.; Abadía, J.; López-Millán, A.-F.; Schmidt, W. Mutually exclusive alterations in secondary metabolism are critical for the uptake of insoluble iron compounds by Arabidopsis and Medicago truncatula. Plant Physiol. 2013, 162, 1473–1485. [Google Scholar] [CrossRef]
- López-Millán, A.-F.; Grusak, M.A.; Abadía, A.; Abadía, J. Iron deficiency in plants: An insight from proteomic approaches. Front. Plant Sci. 2013, 4, 254. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.A.; McCabe, C.E.; Graham, M.A. Dynamic gene expression changes in response to micronutrient, macronutrient, and multiple stress exposures in soybean. Funct. Integr. Genom. 2019, 20, 321–341. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2014, 43, D447–D452. [Google Scholar] [CrossRef]
- Batzenschlager, M.; Schmit, A.-C.; Herzog, E.; Fuchs, J.; Schubert, V.; Houlné, G.; Chabouté, M.-E.J.N. MGO3 and GIP1 act synergistically for the maintenance of centromeric cohesion. Nucleus 2017, 8, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzezinka, K.; Altmann, S.; Bäurle, I. BRUSHY1/TONSOKU/MGOUN3 is required for heat stress memory. Plant Cell Environ. 2018, 42, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Conrath, U.; Beckers, G.J.; Langenbach, C.J.; Jaskiewicz, M.R. Priming for enhanced defense. Annu. Rev. Phytopathol. 2015, 53, 97–119. [Google Scholar] [CrossRef] [PubMed]
- Vriet, C.; Hennig, L.; Laloi, C. Stress-induced chromatin changes in plants: Of memories, metabolites and crop improvement. Cell. Mol. Life Sci. 2015, 72, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.B.; Zilberman, D.J.E. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. Elife 2017, 6, e30674. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.H.; Richards, E.J.; Chung, K.M.; Woo, H.R. Arabidopsis VIM proteins regulate epigenetic silencing by modulating DNA methylation and histone modification in cooperation with MET1. Mol. Plant 2014, 7, 1470–1485. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, T.; Hasegawa, J.; White, C.I.; Matsunaga, S. RAD 54 forms DNA repair foci in response to DNA damage in living plant cells. Plant J. 2017, 90, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, A.; Livanos, P.; Lipka, E.; Gadeyne, A.; Hauser, M.T.; Van Damme, D.; Müller, S. Dual localized kinesin-12 POK2 plays multiple roles during cell division and interacts with MAP65-3. EMBO Rep. 2018, 19, e46085. [Google Scholar] [CrossRef]
- Mittler, R.; Kim, Y.; Song, L.; Coutu, J.; Coutu, A.; Ciftci-Yilmaz, S.; Lee, H.; Stevenson, B.; Zhu, J.-K. Gain-and loss-of-function mutations in Zat10 enhance the tolerance of plants to abiotic stress. FEBS Lett. 2006, 580, 6537–6542. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-M.; An, J.; Han, H.J.; Kim, S.H.; Lim, C.O.; Yun, D.-J.; Chung, W.S. ZAT11, a zinc finger transcription factor, is a negative regulator of nickel ion tolerance in Arabidopsis. Plant Cell Rep. 2014, 33, 2015–2021. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Gao, Y.; Jones, A.M. Extra large G-protein interactome reveals multiple stress response function and partner-dependent XLG subcellular localization. Front. Plant Sci. 2017, 8, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Grubb, L.E.; Wang, J.; Liang, X.; Li, L.; Gao, C.; Ma, M.; Feng, F.; Li, M.; Li, L. A regulatory module controlling homeostasis of a plant immune kinase. Mol. Cell 2018, 69, 493–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grusak, M.A.; Pezeshgi, S. Shoot-to-root signal transmission regulates root Fe (III) reductase activity in the dgl mutant of pea. Plant Physiol. 1996, 110, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Schikora, A.; Schmidt, W. Iron stress-induced changes in root epidermal cell fate are regulated independently from physiological responses to low iron availability. Plant Physiol. 2001, 125, 1679–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, Y.; Hodoshima, H.; Shimada, H.; Shoji, K.; Yoshihara, T.; Goto, F. Long-distance signals positively regulate the expression of iron uptake genes in tobacco roots. Planta 2007, 227, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Garnica, M.; Bacaicoa, E.; Mora, V.; San Francisco, S.; Baigorri, R.; Zamarreño, A.M.; Garcia-Mina, J.M. Shoot iron status and auxin are involved in iron deficiency-induced phytosiderophores release in wheat. BMC Plant Biol. 2018, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, G.; Chen, P.; Zhu, H.; Wang, S.; Ding, Y. Shoot-root communication plays a key role in physiological alterations of rice (Oryza sativa) under iron deficiency. Front. Plant Sci. 2018, 9, 757. [Google Scholar] [CrossRef]
- Lin, S.-F.; Baumer, J.S.; Ivers, D.; de Cianzo, S.R.; Shoemaker, R.C. Field and nutrient solution tests measure similar mechanisms controlling iron deficiency chlorosis in soybean. Crop. Sci. 1998, 38, 254–259. [Google Scholar] [CrossRef]
- Lin, S.F.; Baumer, J.; Ivers, D.; Cianzio, S.; Shoemaker, R. Nutrient solution screening of Fe chlorosis resistance in soybean evaluated by molecular characterization. J. Plant Nutr. 2000, 23, 1915–1928. [Google Scholar] [CrossRef]
- Lin, S.F.; Grant, D.; Cianzio, S.; Shoemaker, R. Molecular characterization of iron deficiency chlorosis in soybean. J. Plant Nutr. 2000, 23, 1929–1939. [Google Scholar] [CrossRef]
- Severin, A.J.; Peiffer, G.A.; Xu, W.W.; Hyten, D.L.; Bucciarelli, B.; O’Rourke, J.A.; Bolon, Y.-T.; Grant, D.; Farmer, A.D.; May, G.D. An integrative approach to genomic introgression mapping. Plant Physiol. 2010, 154, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, A.O.; Bhaskar, P.B.; Bolon, Y.T.; Nolan, R.; Shoemaker, R.C.; Vance, C.P.; Stupar, R.M. Genomic heterogeneity and structural variation in soybean near isogenic lines. Front. Plant Sci. 2013, 4, 104. [Google Scholar] [CrossRef] [Green Version]
- Assefa, T.; Zhang, J.; Chowda-Reddy, R.; Lauter, A.N.M.; Singh, A.; O’Rourke, J.A.; Graham, M.A.; Singh, A.K. Deconstructing the genetic architecture of iron deficiency chlorosis in soybean using genome-wide approaches. BMC Plant Biol. 2020, 20, 42. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, T. Speciation and cytogenetics. In Soybeans: Improvement, Production and Uses, 3rd ed.; Boerma, H.R., Specht, J.E., Eds.; American Society of Agronomy, Crop Science Society of America, Soil Science Society of America: Madison, WI, USA, 2004; pp. 97–129. [Google Scholar]
- Rincker, K.; Nelson, R.; Specht, J.; Sleper, D.; Cary, T.; Cianzio, S.R.; Casteel, S.; Conley, S.; Chen, P.; Davis, V.; et al. Genetic improvement of U.S. Soybean in maturity groups II, III and IV. Crop. Sci. 2014, 54, 1419–1432. [Google Scholar] [CrossRef]
- Johnson, H.W. Registration of soybean varities, VI. Agron. J. 1958, 50, 690. [Google Scholar] [CrossRef]
- Germplasm Resources Information Network (GRIN) National Genetic Resources Program. Available online: https://www.ars-grin.gov (accessed on 19 May 2020).
- Chaney, R.L.; Coulombe, B.A.; Bell, P.F.; Angle, J.S. Detailed method to screen dicot cultivars for resistance to Fe-chlorosis using FeDTPA and bicarbonate in nutrient solutions. J. Plant Nutr. 1992, 15, 2063–2083. [Google Scholar] [CrossRef]
- Buffalo, V. Scythe-a Bayesian Adaptor Trimmer. Version 0.994 BETA. Available online: https://github.com/vsbuffalo/scythe (accessed on 19 May 2020).
- The FASTX-Toolkit. FASTQ/A Short-Reads Pre-Processing Tools. Version 0.0.13. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 19 May 2020).
- Sickle-A Windowed Adaptive Trimming Tool for FASTQ Files Using Quality. Version 1.33. Available online: https://github.com/najoshi/sickle (accessed on 19 May 2020).
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Morgan, M.; Pages, H. Rsamtools: Binary Alignment (BAM), Variant Call (BCF), or Tabix File Import (R Package Version 1.12.4); R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pages, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Smyth, G.K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 2007, 23, 2881–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Lindsay, H.; Robinson, M.D. Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 2014, 42, e91. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant graphics for data analysis. In Use R! Springer: New York, NY, USA, 2009. [Google Scholar]
- Rutter, L.; Moran Lauter, A.N.; Graham, M.A.; Cook, D. Visualization methods for differential expression analysis. BMC Bioinform. 2019, 20, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; Smyth, G.K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 2007, 9, 321–332. [Google Scholar] [CrossRef] [Green Version]
- The SoyBase Genome Annotation Report Page. Version Wm82.a2.v2. Available online: http://soybase.org/genomeannotation.index.php (accessed on 19 May 2020).
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Murtagh, F. A survey of algorithms for contiguity-constrained clustering and related problems. Comput. J. 1985, 28, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Murtagh, F.; Legendre, P. Ward’s hierarchical agglomerative clustering method: Which algorithms implement ward’s criterion? J. Class. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- The SoyBase GO Term Enrichment Tool. Version Wm82.a2.v2. Available online: https://www.soybase.org/goslimgraphic_v2/dashboard.php (accessed on 19 May 2020).
- Morales, A.M.A.P.; O’Rourke, J.A.; van de Mortel, M.; Scheider, K.T.; Bancroft, T.J.; Borem, A.; Nelson, R.T.; Nettleton, D.; Baum, T.J.; Shoemaker, R.C.; et al. Transcriptome analyses and virus induced gene silencing identify genes in the Rpp4-mediated Asian soybean rust resistance pathway. Funct. Plant Biol. 2013, 40, 1029–1047. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | GO Term ID | GO Term Description | Genome Count | DEG Count | Corrected p-Value |
|---|---|---|---|---|---|
| L1 | GO:0008283 | Cell proliferation | 388 | 161 | 5.64 × 10−113 |
| L1 | GO:0006275 | Regulation of DNA replication | 255 | 124 | 3.41 × 10−97 |
| L1 | GO:0006260 | DNA replication | 270 | 121 | 2.05 × 10−89 |
| L1 | GO:0051567 | Histone H3-K9 methylation | 443 | 148 | 7.86 × 10−88 |
| L1 | GO:0006270 | DNA replication initiation | 159 | 91 | 1.45 × 10−79 |
| L1 | GO:0000911 | Cytokinesis by cell plate formation | 471 | 139 | 2.16 × 10−74 |
| L1 | GO:0010389 | Regulation of G2/M transition of mitotic cell cycle | 155 | 85 | 4.88 × 10−72 |
| L1 | GO:0006306 | DNA methylation | 421 | 124 | 4.27 × 10−66 |
| L1 | GO:0051726 | Regulation of cell cycle | 345 | 113 | 1.19 × 10−65 |
| L1 | GO:0006334 | Nucleosome assembly | 128 | 73 | 2.18 × 10−63 |
| L2 | GO:0009658 | Chloroplast organization | 316 | 31 | 1.09 × 10−13 |
| L2 | GO:0006364 | rRNA processing | 532 | 38 | 1.71 × 10−12 |
| L2 | GO:0019288 | Isopentenyl diphosphate biosynthetic process, mevalonat x 10-independent pathway | 581 | 39 | 5.56 × 10−12 |
| L2 | GO:0045036 | Protein targeting to chloroplast | 114 | 18 | 6.05 × 10−11 |
| L2 | GO:0042793 | Transcription from plastid promoter | 169 | 21 | 7.32 × 10−11 |
| L2 | GO:0010027 | Thylakoid membrane organization | 469 | 29 | 9.76 × 10−8 |
| L2 | GO:0016226 | Iron-sulfur cluster assembly | 227 | 19 | 1.02 × 10−6 |
| L2 | GO:0045893 | Positive regulation of transcription, DNA-dependent | 1080 | 41 | 3.97 × 10−5 |
| L2 | GO:0009902 | Chloroplast relocation | 230 | 16 | 2.44 × 10−4 |
| L2 | GO:0006655 | Phosphatidylglycerol biosynthetic process | 176 | 14 | 2.56 × 10−4 |
| L3 | GO:0048767 | Root hair elongation | 504 | 31 | 1.30 × 10−5 |
| L3 | GO:0016126 | Sterol biosynthetic process | 363 | 23 | 4.87 × 10−4 |
| L3 | GO:0006949 | Syncytium formation | 63 | 9 | 1.92 × 10−3 |
| L3 | GO:0009739 | Response to gibberellin stimulus | 250 | 17 | 5.06 × 10−3 |
| L3 | GO:0006816 | Calcium ion transport | 324 | 19 | 1.25 × 10−2 |
| L3 | GO:0005975 | Carbohydrate metabolic process | 911 | 37 | 1.62 × 10−2 |
| L3 | GO:0000038 | Very long-chain fatty acid metabolic process | 104 | 10 | 2.00 × 10−2 |
| L4 | GO:0006878 | Cellular copper ion homeostasis | 17 | 5 | 2.19 × 10−3 |
| L4 | GO:0000103 | Sulfate assimilation | 32 | 6 | 3.91 × 10−3 |
| L4 | GO:0055085 | Transmembrane transport | 1061 | 36 | 4.39 × 10−3 |
| Cluster | GO Term ID | GO Term Description | Genome Count | DEG Count | Corrected p-Value |
|---|---|---|---|---|---|
| R1 | GO:0043069 | Negative regulation of programmed cell death | 525 | 76 | 7.92 × 10−14 |
| R1 | GO:0002679 | Respiratory burst involved in defense response | 420 | 61 | 6.99 × 10−11 |
| R1 | GO:0031348 | Negative regulation of defense response | 766 | 89 | 1.52 × 10−10 |
| R1 | GO:0009863 | Salicylic acid mediated signaling pathway | 458 | 62 | 1.04 × 10−9 |
| R1 | GO:0050832 | Defense response to fungus | 941 | 100 | 1.40 × 10−9 |
| R1 | GO:0000165 | MAPK cascade | 575 | 70 | 6.40 × 10−9 |
| R1 | GO:0010363 | Regulation of plant-type hypersensitive response | 1019 | 103 | 1.23 × 10−8 |
| R1 | GO:0009862 | Systemic acquired resistance, salicylic acid mediated | 688 | 77 | 4.19 × 10−8 |
| R1 | GO:0006612 | Protein targeting to membrane | 1020 | 101 | 6.11 × 10−8 |
| R1 | GO:0009697 | Salicylic acid biosynthetic process | 653 | 73 | 1.50 × 10−7 |
| R2 | GO:0006412 | Translation | 763 | 100 | 6.94 × 10−29 |
| R2 | GO:0042254 | Ribosome biogenesis | 264 | 37 | 9.55 × 10−11 |
| R2 | GO:0001510 | RNA methylation | 418 | 46 | 8.74 x 10−10 |
| R2 | GO:0009664 | Plant-type cell wall organization | 337 | 39 | 9.48 × 10−9 |
| R2 | GO:0009834 | Secondary cell wall biogenesis | 135 | 21 | 2.61 × 10−6 |
| R2 | GO:0044036 | Cell wall macromolecule metabolic process | 222 | 27 | 4.35 × 10−6 |
| R2 | GO:0015979 | Photosynthesis | 452 | 40 | 1.44 × 10−5 |
| R2 | GO:0010089 | Xylem development | 225 | 26 | 2.39 × 10−5 |
| R2 | GO:0010014 | Meristem initiation | 342 | 33 | 3.20 × 10−5 |
| R2 | GO:0006949 | Syncytium formation | 63 | 13 | 8.33 × 10−5 |
| R3 | GO:0006412 | Translation | 763 | 110 | 2.38 × 10−36 |
| R3 | GO:0009834 | Secondary cell wall biogenesis | 135 | 46 | 2.17 × 10−31 |
| R3 | GO:0042254 | Ribosome biogenesis | 264 | 53 | 9.57 × 10−24 |
| R3 | GO:0001510 | RNA methylation | 418 | 58 | 1.03 × 10−17 |
| R3 | GO:0015979 | Photosynthesis | 452 | 54 | 1.66 × 10−13 |
| R3 | GO:0045492 | Xylan biosynthetic process | 497 | 56 | 6.24 × 10−13 |
| R3 | GO:0010207 | Photosystem II assembly | 372 | 46 | 7.35 × 10−12 |
| R3 | GO:0006364 | RNA processing | 532 | 56 | 1.16 × 10−11 |
| R3 | GO:0019684 | Photosynthesis, light reaction | 320 | 42 | 1.33 × 10−11 |
| R3 | GO:0009773 | Photosynthetic electron transport, photosystem I | 121 | 25 | 8.97 × 10−11 |
| R4 | GO:0006487 | Protein N-linked glycosylation | 298 | 40 | 2.65 × 10−6 |
| R4 | GO:0015706 | Nitrate transport | 486 | 54 | 7.70 × 10−6 |
| R4 | GO:0006468 | Protein phosphorylation | 2386 | 171 | 5.38 × 10−5 |
| R4 | GO:0009863 | Salicylic acid mediated signaling pathway | 458 | 49 | 9.58 × 10−5 |
| R4 | GO:0045087 | Innate immune response | 274 | 34 | 2.69 × 10−4 |
| R4 | GO:0006952 | Defense response | 1116 | 91 | 3.78 × 10−4 |
| R4 | GO:0010106 | Cellular response to iron ion starvation | 237 | 30 | 8.67 × 10−4 |
| R4 | GO:0071366 | Cellular response to indolebutyric acid stimulus | 10 | 6 | 1.98 × 10−3 |
| R4 | GO:0030968 | Endoplasmic reticulum unfolded protein response | 501 | 49 | 2.04 × 10−3 |
| R4 | GO:0000041 | Transition metal ion transport | 201 | 25 | 9.36 × 10−3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauter, A.N.M.; Rutter, L.; Cook, D.; O’Rourke, J.A.; Graham, M.A. Examining Short-Term Responses to a Long-Term Problem: RNA-Seq Analyses of Iron Deficiency Chlorosis Tolerant Soybean. Int. J. Mol. Sci. 2020, 21, 3591. https://doi.org/10.3390/ijms21103591
Lauter ANM, Rutter L, Cook D, O’Rourke JA, Graham MA. Examining Short-Term Responses to a Long-Term Problem: RNA-Seq Analyses of Iron Deficiency Chlorosis Tolerant Soybean. International Journal of Molecular Sciences. 2020; 21(10):3591. https://doi.org/10.3390/ijms21103591
Chicago/Turabian StyleLauter, Adrienne N. Moran, Lindsay Rutter, Dianne Cook, Jamie A. O’Rourke, and Michelle A. Graham. 2020. "Examining Short-Term Responses to a Long-Term Problem: RNA-Seq Analyses of Iron Deficiency Chlorosis Tolerant Soybean" International Journal of Molecular Sciences 21, no. 10: 3591. https://doi.org/10.3390/ijms21103591
APA StyleLauter, A. N. M., Rutter, L., Cook, D., O’Rourke, J. A., & Graham, M. A. (2020). Examining Short-Term Responses to a Long-Term Problem: RNA-Seq Analyses of Iron Deficiency Chlorosis Tolerant Soybean. International Journal of Molecular Sciences, 21(10), 3591. https://doi.org/10.3390/ijms21103591