Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons


Abstract
:1. Introduction
1.1. Diabetes and Neurodegeneration
1.2. Diabetes and NAD+
1.3. Diabetes, SIRT1, and NAD+
2. Results
2.1. NMN Normalized Diabetes-Induced Decrease in Brain NAD+ Levels
2.2. NMN Normalized Diabetes-Induced Changes in Brain Biochemicals
2.3. NMN Administration Prevents Diabetes-Induced Hippocampal Changes
2.3.1. NMN Administration Prevents Loss of Hippocampal CA1 Volume
2.3.2. NMN Administration Prevents a Loss of Hippocampal CA1 Neurons
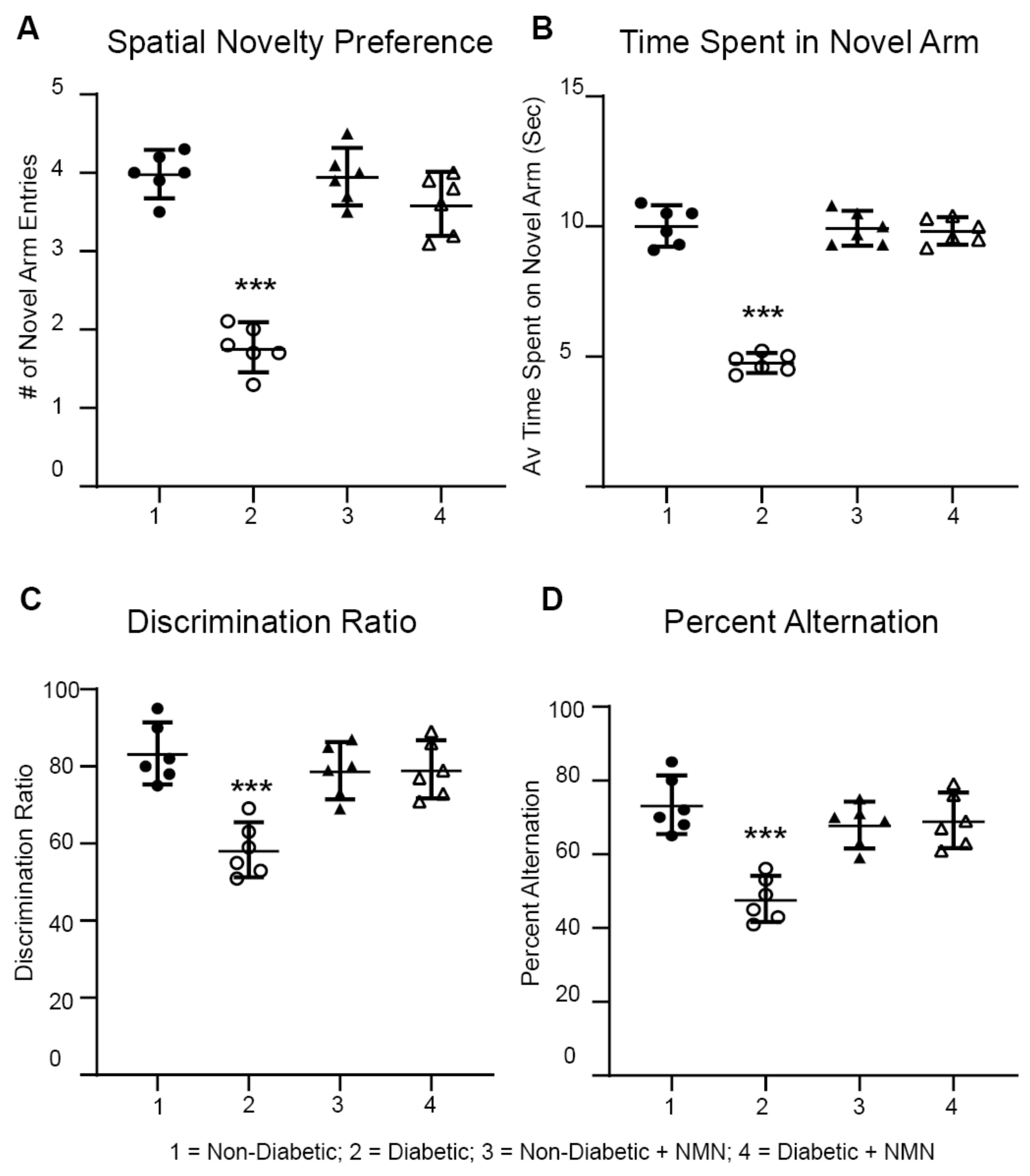
2.4. NMN Administration Prevents Diabetes-Induced Memory Impairment
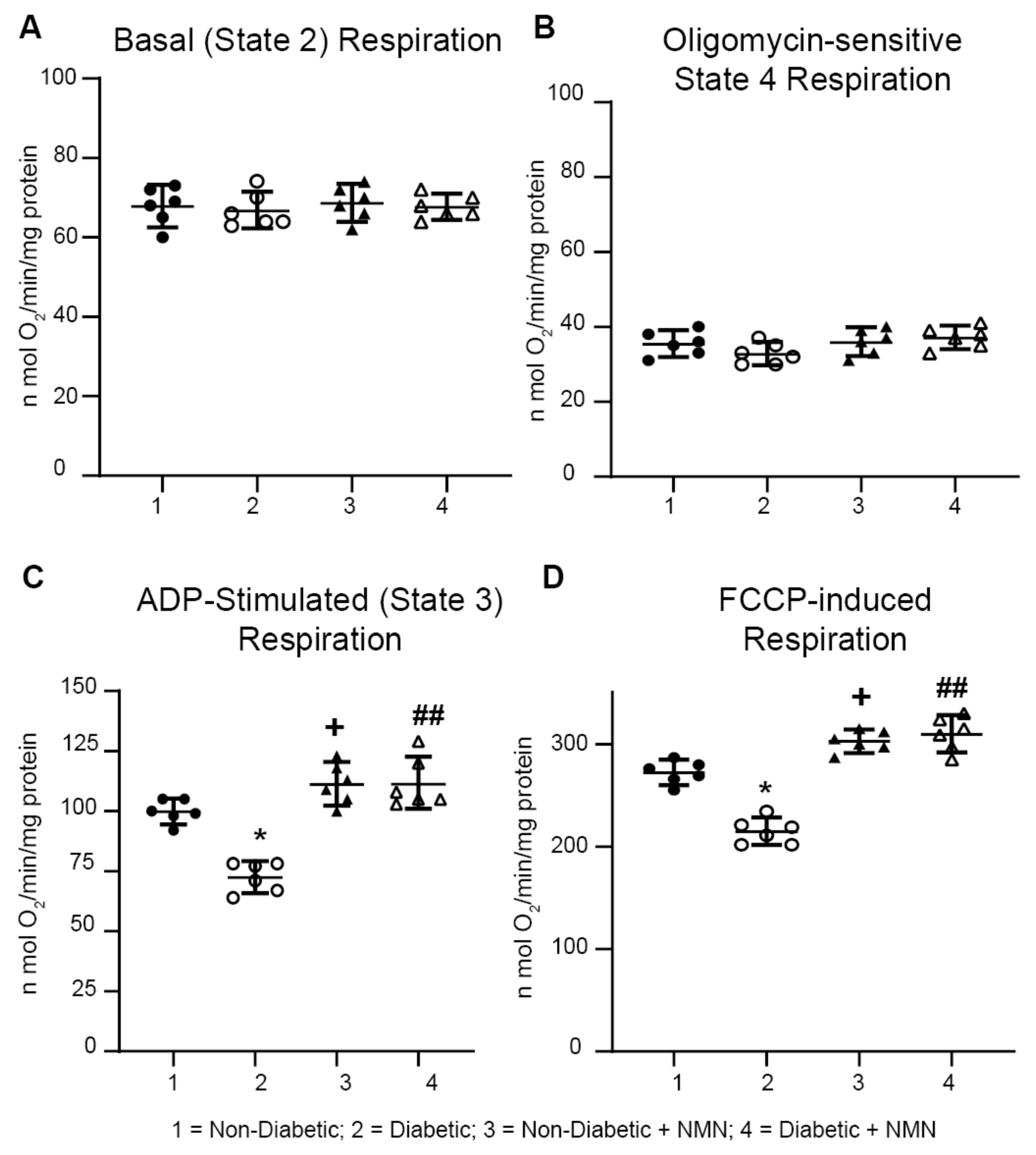
2.5. NMN Administration Increases Mitochondrial Spare Reserve Capacity
2.6. NMN Administration Upregulates the NAD+-Dependent SIRT1 Signaling Pathway and Downregulates the Acetylation Pathway in the Hippocampus of Diabetic Rats
3. Discussion
3.1. Diabetes and NMN
3.2. The NAD+ Precursor NMN as a Therapeutic Agent
3.3. Diabetes-Induced Changes in Biochemicals is Prevented by NMN Administration
3.4. NMN Prevents Diabetes-Induced Decreases in Hippocampal Volume and Neuronal Counts
3.5. Diabetes-Induced Decreases in Mitochondrial Respiration was Rescued by NMN Administration
3.6. NMN Administration Increases SIRT1 and Deacetylation of Proteins
4. Materials and Methods
4.1. Animal Procedure
4.2. Quantification of NAD+
4.3. Magnetic Resonance Imaging (MRI) and Magnetic Resonance Spectroscopy (MRS)
4.4. Tissue Processing
4.5. Estimation of the Volume of Hippocampus, CA1, and DG
4.6. Estimation of Total Neuronal Numbers in CA1 and DG Regions of the Hippocampus
4.7. Behavioral Memory Tests
4.8. Isolation of Mitochondria
4.9. Mitochondrial Respiration
4.10. In-Gel Extraction and Mass Spectroscopy Analysis of Major Acetylated Protein
4.11. Western Blot Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grunblatt, E.; Bartl, J.; Riederer, P. The link between iron, metabolic syndrome, and Alzheimer’s disease. J. Neural Transm. 2011, 118, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sun, Z.; Han, X.; Li, S.; Jiang, X.; Chen, S.; Zhang, J.; Lu, H. Neuroprotective effect of resveratrol via activation of Sirt1 signaling in a rat model of combined diabetes and Alzheimer’s disease. Front. Neurosci. 2019, 13, 1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lian, S.; Zhang, J.; Lin, D.; Huang, C.; Liu, L.; Chen, Z. Mitochondrial perturbation contributing to Cognitive Decline in Streptozotocin-Induced Type 1 Diabetic Rats. Cell Physiol. Biochem. 2018, 46, 1668–1682. [Google Scholar] [CrossRef]
- Cardoso, S.; Santos, M.S.; Seica, R.; Moreira, P.I. Cortical and hippocampal mitochondria bioenergetics and oxidative status during hyperglycemia and/or insulin-induced hypoglycemia. Biochim. Biophys. Acta 2010, 1802, 942–951. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Santos, M.S.; Moreno, A.M.; Proenca, T.; Seica, R.; Oliveira, C.R. Effect of streptozotocin-induced diabetes on rat brain mitochondria. J. Neuroendocrinol. 2004, 16, 32–38. [Google Scholar] [CrossRef]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [Green Version]
- Ortiz Mdel, C.; Lores-Arnaiz, S.; Albertoni Borghese, M.F.; Balonga, S.; Lavagna, A.; Filipuzzi, A.L.; Cicerchia, D.; Majowicz, M.; Bustamante, J. Mitochondrial dysfunction in brain cortex mitochondria of STZ-diabetic rats: Effect of l-Arginine. Neurochem. Res. 2013, 38, 2570–2580. [Google Scholar] [CrossRef]
- Osorio-Paz, I.; Ramirez-Perez, G.; Hernandez-Ramirez, J.E.; Uribe-Carvajal, S.; Salceda, R. Mitochondrial activity in different regions of the brain at the onset of streptozotocin-induced diabetes in rats. Mol. Biol. Rep. 2018, 45, 871–879. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim. Biophys. Acta 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Huang, M.; Gao, L.; Lei, H. Region-specific cerebral metabolic alterations in streptozotocin-induced type 1 diabetic rats: An in vivo proton magnetic resonance spectroscopy study. J. Cereb. Blood Flow Metab. 2015, 35, 1738–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Wang, G.; Han, S.S.; He, M.Y.; Cheng, X.; Ma, Z.L.; Wu, X.; Yang, X.; Liu, G.S. Effects of oxidative stress on hyperglycaemia-induced brain malformations in a diabetes mouse model. Exp. Cell Res. 2016, 347, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fan, S.; Song, D.; Wang, Z.; Ma, S.; Li, S.; Li, X.; Xu, M.; Xu, M.; Wang, X. Long-term streptozotocin-induced diabetes in rats leads to severe damage of brain blood vessels and neurons via enhanced oxidative stress. Mol. Med. Rep. 2013, 7, 431–440. [Google Scholar] [CrossRef]
- Zhao, F.; Li, J.; Mo, L.; Tan, M.; Zhang, T.; Tang, Y.; Zhao, Y. Changes in neurons and synapses in hippocampus of streptozotocin-induced type 1 diabetes rats: A stereological investigation. Anat. Rec. 2016, 299, 1174–1183. [Google Scholar] [CrossRef] [Green Version]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in aging: Molecular mechanisms and translational implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) intermediates: The biology and therapeutic potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD(+) metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Osterloh, J.M.; Yang, J.; Rooney, T.M.; Fox, A.N.; Adalbert, R.; Powell, E.H.; Sheehan, A.E.; Avery, M.A.; Hackett, R.; Logan, M.A.; et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 2012, 337, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef] [Green Version]
- Turkiew, E.; Falconer, D.; Reed, N.; Hoke, A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J. Peripher. Nerv. Syst. 2017, 22, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, Y. Metabolic aspects of neuronal degeneration: From a NAD(+) point of view. Neurosci. Res. 2019, 139, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008, 582, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Dominy, J.E., Jr.; Lee, Y.; Gerhart-Hines, Z.; Puigserver, P. Nutrient-dependent regulation of PGC-1alpha’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim. Biophys. Acta 2010, 1804, 1676–1683. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Tanner, K.G.; Landry, J.; Sternglanz, R.; Denu, J.M. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA 2000, 97, 14178–14182. [Google Scholar] [PubMed] [Green Version]
- Jackson, M.D.; Schmidt, M.T.; Oppenheimer, N.J.; Denu, J.M. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. [Google Scholar] [CrossRef] [Green Version]
- Chalkiadaki, A.; Guarente, L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat. Rev. Endocrinol. 2012, 8, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Choi, J.; Chandrasekaran, K.; Demarest, T.G.; Kristian, T.; Xu, S.; Vijaykumar, K.; Dsouza, K.G.; Qi, N.R.; Yarowsky, P.J.; Gallipoli, R.; et al. Brain diabetic neurodegeneration segregates with low intrinsic aerobic capacity. Ann. Clin. Transl. Neurol. 2014, 1, 589–604. [Google Scholar] [CrossRef] [Green Version]
- Wiegers, E.C.; Rooijackers, H.M.; van Asten, J.J.A.; Tack, C.J.; Heerschap, A.; de Galan, B.E.; van der Graaf, M. Elevated brain glutamate levels in type 1 diabetes: Correlations with glycaemic control and age of disease onset but not with hypoglycaemia awareness status. Diabetologia 2019, 62, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Dong, M.; Ren, M.; Li, C.; Zheng, H.; Gao, H. Metabolomic analysis identifies lactate as an important pathogenic factor in diabetes-associated cognitive decline rats. Mol. Cell Proteomics 2018, 17, 2335–2346. [Google Scholar] [CrossRef] [Green Version]
- Golub, V.M.; Brewer, J.; Wu, X.; Kuruba, R.; Short, J.; Manchi, M.; Swonke, M.; Younus, I.; Reddy, D.S. Neurostereology protocol for unbiased quantification of neuronal injury and neurodegeneration. Front. Aging Neurosci. 2015, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Slomianka, L.; Gundersen, H.J. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991, 231, 482–497. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Chen, C.; Tu, Y.; Sun, H.T.; Zhao, M.L.; Cheng, S.X.; Qu, Y.; Zhang, S. Sirt1 promotes axonogenesis by deacetylation of Akt and inactivation of GSK3. Mol. Neurobiol. 2013, 48, 490–499. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Salimian, M.; Konduru, S.R.; Choi, J.; Kumar, P.; Long, A.; Klimova, N.; Ho, C.Y.; Kristian, T.; Russell, J.W. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 2019, 142, 3737–3752. [Google Scholar] [CrossRef]
- Roy Chowdhury, S.K.; Smith, D.R.; Saleh, A.; Schapansky, J.; Marquez, A.; Gomes, S.; Akude, E.; Morrow, D.; Calcutt, N.A.; Fernyhough, P. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain 2012, 135, 1751–1766. [Google Scholar] [CrossRef]
- Klimova, N.; Fearnow, A.; Long, A.; Kristian, T. NAD(+) precursor modulates post-ischemic mitochondrial fragmentation and reactive oxygen species generation via SIRT3 dependent mechanisms. Exp. Neurol. 2020, 325, 113144. [Google Scholar] [CrossRef]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.B.; Kubota, S.; Ban, N.; Yoshida, M.; Santeford, A.; Sene, A.; Nakamura, R.; Zapata, N.; Kubota, M.; Tsubota, K.; et al. NAMPT-mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. 2016, 17, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Klimova, N.; Kristian, T. Multi-targeted effect of nicotinamide mononucleotide on brain bioenergetic metabolism. Neurochem. Res. 2019, 44, 2280–2287. [Google Scholar] [CrossRef]
- Long, A.N.; Owens, K.; Schlappal, A.E.; Kristian, T.; Fishman, P.S.; Schuh, R.A. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. 2015, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Mittal, K.; Katare, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. 2016, 10, S144–S149. [Google Scholar] [CrossRef]
- Van Bussel, F.C.; Backes, W.H.; Hofman, P.A.; Puts, N.A.; Edden, R.A.; van Boxtel, M.P.; Schram, M.T.; Stehouwer, C.D.; Wildberger, J.E.; Jansen, J.F. Increased GABA concentrations in type 2 diabetes mellitus are related to lower cognitive functioning. Medicine 2016, 95, e4803. [Google Scholar] [CrossRef]
- Dugan, L.L.; Choi, D.W. Excitotoxicity, free radicals, and cell membrane changes. Ann. Neurol. 1994, 35 (Suppl. 1), S17–S21. [Google Scholar] [CrossRef]
- Alkadhi, K.A. Cellular and molecular differences between area CA1 and the dentate gyrus of the hippocampus. Mol. Neurobiol. 2019, 56, 6566–6580. [Google Scholar] [CrossRef]
- Van Gelder, N.M.; Sherwin, A.L. Metabolic parameters of epilepsy: Adjuncts to established antiepileptic drug therapy. Neurochem. Res. 2003, 28, 353–365. [Google Scholar] [CrossRef]
- Martinez, M.A.; Florenzano, N.V.; Macchia, E.A. Metabolism of N-acetyl-L-aspartate: Its diagnostic and prognostic value. Rev. Neurol. 2016, 62, 361–370. [Google Scholar] [PubMed]
- Moffett, J.R.; Arun, P.; Ariyannur, P.S.; Namboodiri, A.M. N-Acetylaspartate reductions in brain injury: Impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation. Front. Neuroenergetics 2013, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Geddes, D.M.; LaPlaca, M.C.; Cargill, R.S. Susceptibility of hippocampal neurons to mechanically induced injury. Exp. Neurol. 2003, 184, 420–427. [Google Scholar] [CrossRef]
- Lin, B.; Ginsberg, M.D.; Busto, R. Hyperglycemic exacerbation of neuronal damage following forebrain ischemia: Microglial, astrocytic and endothelial alterations. Acta Neuropathol. 1998, 96, 610–620. [Google Scholar] [CrossRef]
- Eichenbaum, H. Hippocampus: Mapping or memory? Curr. Biol. 2000, 10, R785–R787. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.; Sommers, M.S.; Lucki, I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neurosci. Biobehav. Rev. 2013, 37, 1346–1362. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, F.; Gagnon, S.; Massicotte, G. Hippocampal synaptic plasticity and glutamate receptor regulation: Influences of diabetes mellitus. Eur. J. Pharmacol. 2004, 490, 177–186. [Google Scholar] [CrossRef]
- Sansbury, B.E.; Jones, S.P.; Riggs, D.W.; Darley-Usmar, V.M.; Hill, B.G. Bioenergetic function in cardiovascular cells: The importance of the reserve capacity and its biological regulation. Chem. Biol. Interact. 2011, 191, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 mediates metabolic homeostasis and neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- Sun, A.; Wei, J.; Childress, C.; Shaw, J.H.t.; Peng, K.; Shao, G.; Yang, W.; Lin, Q. The E3 ubiquitin ligase NEDD4 is an LC3-interactive protein and regulates autophagy. Autophagy 2017, 13, 522–537. [Google Scholar] [CrossRef] [Green Version]
- Camera, D.; Coleman, H.A.; Parkington, H.C.; Jenkins, T.A.; Pow, D.V.; Boase, N.; Kumar, S.; Poronnik, P. Learning, memory and long-term potentiation are altered in Nedd4 heterozygous mice. Behav. Brain Res. 2016, 303, 176–181. [Google Scholar] [CrossRef]
- DiAntonio, A. Nedd4 branches out. Neuron 2010, 65, 293–294. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Yang, L.; Qin, A.R.; Ly, J.; Liederer, B.M.; Messick, K.; Ma, S.; Zak, M.; Dragovich, P.S.; Dean, B.J.; et al. Measuring NAD(+) levels in mouse blood and tissue samples via a surrogate matrix approach using LC-MS/MS. Bioanalysis 2014, 6, 1445–1457. [Google Scholar] [CrossRef]
- Kristian, T.; Balan, I.; Schuh, R.; Onken, M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J. Neurosci. Res. 2011, 89, 1946–1955. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Schuh, R.; Kristian, T. Mitochondrial dysfunction and NAD(+) metabolism alterations in the pathophysiology of acute brain injury. Transl. Stroke Res. 2013, 4, 618–634. [Google Scholar] [CrossRef]
- Anjaneyulu, M.; Berent-Spillson, A.; Inoue, T.; Choi, J.; Cherian, K.; Russell, J.W. Transforming growth factor-beta induces cellular injury in experimental diabetic neuropathy. Exp. Neurol. 2008, 211, 469–479. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.W.; Berent-Spillson, A.; Vincent, A.M.; Freimann, C.L.; Sullivan, K.A.; Feldman, E.L. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol. Dis. 2008, 30, 420–429. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Non-Dia (n = 6) 1 | Dia (n = 6) 2 | Non-Dia + NMN (n = 6) 3 | Dia + NMN (n = 6) 4 | 1 vs. 2 | 2 vs. 4 | 1 vs. 3 | 3 vs. 4 | 1 vs. 4 |
|---|---|---|---|---|---|---|---|---|---|
| Body Weight (g) | 416 ± 26 | 318 ± 21 | 421 ± 32 | 296 ± 14 | <0.05 | NS | NS | <0.05 | <0.05 |
| Glucose (mM) | 6.1 ± 0.6 | 22.7 ± 2 | 6 ± 0.4 | 24.6 ± 1.3 | <0.001 | NS | NS | <0.001 | <0.001 |
| Cholesterol (mg/dL) | 172 ± 14 | 166 ± 14 | 165 ± 12 | 163.4 ± 22 | NS | NS | NS | NS | NS |
| Triglycerides (mg/dL) | 185 ± 6 | 120 ± 3 | 177 ± 8 | 124 ± 6 | <0.001 | NS | NS | <0.001 | <0.001 |
| GTT-AUC (mg/dL/min) | 10472 ± 200 | 30030 ± 300 | 9989 ± 190 | 29854 ± 278 | <0.001 | NS | NS | <0.001 | <0.001 |
| Brain NAD+ (pmol/mg) | 200 ± 15 | 123 ± 15 | 237 ± 13 | 198 ± 12 | <0.05 | <0.05 | NS | <0.05 | NS |
| Metabolite mM | Non-Dia 1 | Dia 2 | Non-Dia + NMN 3 | Dia + NMN 4 | 1 vs. 2 | 2 vs. 4 | 1 vs. 3 | 3 vs. 4 | 1 vs. 4 |
|---|---|---|---|---|---|---|---|---|---|
| GABA | 0.29 | 0.33 | 0.27 | 0.27 | 0.018 | 0.01 | NS | NS | NS |
| Glucose | 0.61 | 1.07 | 0.60 | 0.97 | <0.001 | NS | NS | 0.002 | 0.002 |
| Glutamine | 0.38 | 0.37 | 0.36 | 0.36 | NS | NS | NS | NS | NS |
| Glutamate | 1.16 | 1.31 | 1.14 | 1.14 | <0.001 | <0.001 | NS | NS | NS |
| Ph. Choline | 0.14 | 0.12 | 0.14 | 0.15 | NS | NS | NS | NS | NS |
| Myoinositol | 1.00 | 1.15 | 1.00 | 1.16 | 0.018 | NS | NS | NS | 0.014 |
| NAA | 1.22 | 1.03 | 1.23 | 1.19 | 0.002 | 0.003 | NS | NS | NS |
| Taurine | 0.81 | 0.91 | 0.79 | 0.87 | <0.001 | 0.010 | NS | 0.04 | 0.007 |
| GSH | 0.25 | 0.17 | 0.27 | 0.21 | <0.001 | 0.006 | NS | 0.03 | 0.009 |
| GPC + PCh | 0.18 | 0.17 | 0.17 | 0.19 | NS | NS | NS | NS | NS |
| NAA + NAAG | 1.17 | 1.19 | 1.14 | 1.14 | NS | 0.013 | NS | NS | NS |
| Glu + Gln | 1.54 | 1.67 | 1.51 | 1.50 | 0.031 | 0.008 | NS | NS | NS |
| nmol O2 /min/mg Protein | Non-Dia 1 | Dia 2 | Non-Dia + NMN 3 | Dia + NMN 4 | 1 vs. 2 | 2 vs. 4 | 1 vs. 3 | 3 vs. 4 | 1 vs. 4 |
|---|---|---|---|---|---|---|---|---|---|
| State 2 Respiration | 63.8 ± 4.8 | 68.6 ± 4.3 | 66.8 ± 4.3 | 69.6 ± 2.9 | NS | NS | NS | NS | NS |
| State 3 Respiration | 101.8 ± 5.3 | 82.2 ± 11.8 | 119.8 ± 7.5 | 109.3 ± 5.6 | <0.05 | <0.01 | <0.05 | NS | NS |
| State 4 Respiration | 35.5 ± 3.3 | 36 ± 3.5 | 32.8 ± 2.8 | 37.2 ± 2.9 | NS | NS | NS | NS | NS |
| FCCP-Induced Respiration | 272 ± 11.3 | 240 ± 13.6 | 302.7 ± 10.3 | 295 ± 16.8 | <0.05 | <0.01 | <0.05 | NS | <0.05 |
| Spare Reserve Capacity | 204 ± 15.1 | 162 ± 16 | 235 ± 11 | 242 ± 17.7 | <0.05 | <0.01 | <0.05 | NS | <0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, K.; Choi, J.; Arvas, M.I.; Salimian, M.; Singh, S.; Xu, S.; Gullapalli, R.P.; Kristian, T.; Russell, J.W. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. Int. J. Mol. Sci. 2020, 21, 3756. https://doi.org/10.3390/ijms21113756
Chandrasekaran K, Choi J, Arvas MI, Salimian M, Singh S, Xu S, Gullapalli RP, Kristian T, Russell JW. Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. International Journal of Molecular Sciences. 2020; 21(11):3756. https://doi.org/10.3390/ijms21113756
Chicago/Turabian StyleChandrasekaran, Krish, Joungil Choi, Muhammed Ikbal Arvas, Mohammad Salimian, Sujal Singh, Su Xu, Rao P Gullapalli, Tibor Kristian, and James William Russell. 2020. "Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons" International Journal of Molecular Sciences 21, no. 11: 3756. https://doi.org/10.3390/ijms21113756
APA StyleChandrasekaran, K., Choi, J., Arvas, M. I., Salimian, M., Singh, S., Xu, S., Gullapalli, R. P., Kristian, T., & Russell, J. W. (2020). Nicotinamide Mononucleotide Administration Prevents Experimental Diabetes-Induced Cognitive Impairment and Loss of Hippocampal Neurons. International Journal of Molecular Sciences, 21(11), 3756. https://doi.org/10.3390/ijms21113756