Activation of Mitochondrial 2-Oxoglutarate Dehydrogenase by Cocarboxylase in Human Lung Adenocarcinoma Cells A549 Is p53/p21-Dependent and Impairs Cellular Redox State, Mimicking the Cisplatin Action


 ,
,
Abstract
:
1. Introduction
2. Results
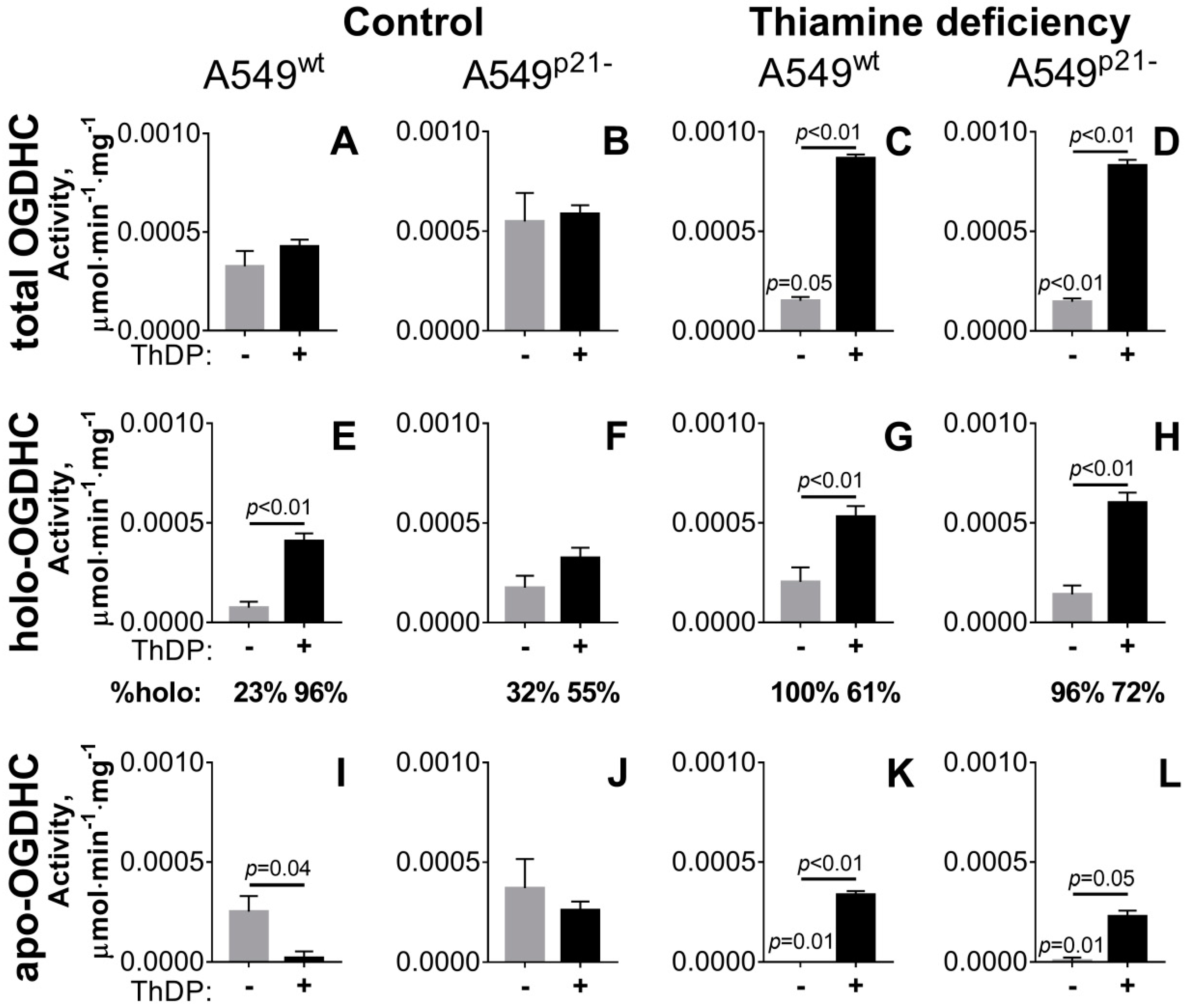
2.1. Incubation of A549 Cells with 5 mM ThDP Saturates the Mitochondrial 2-Oxoglutarate Dehydrogenase with ThDP in a p21-Dependent Manner
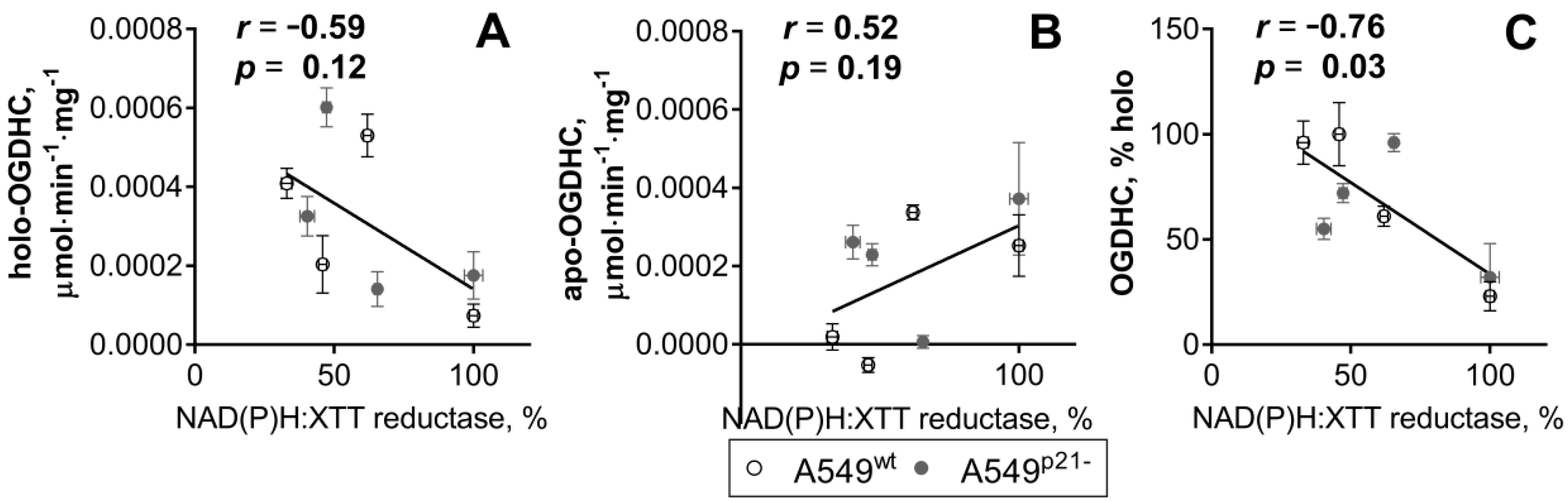
2.2. Knockdown of p21 Deregulates Partition of Total OGDHC into the Apo- and Holoenzyme
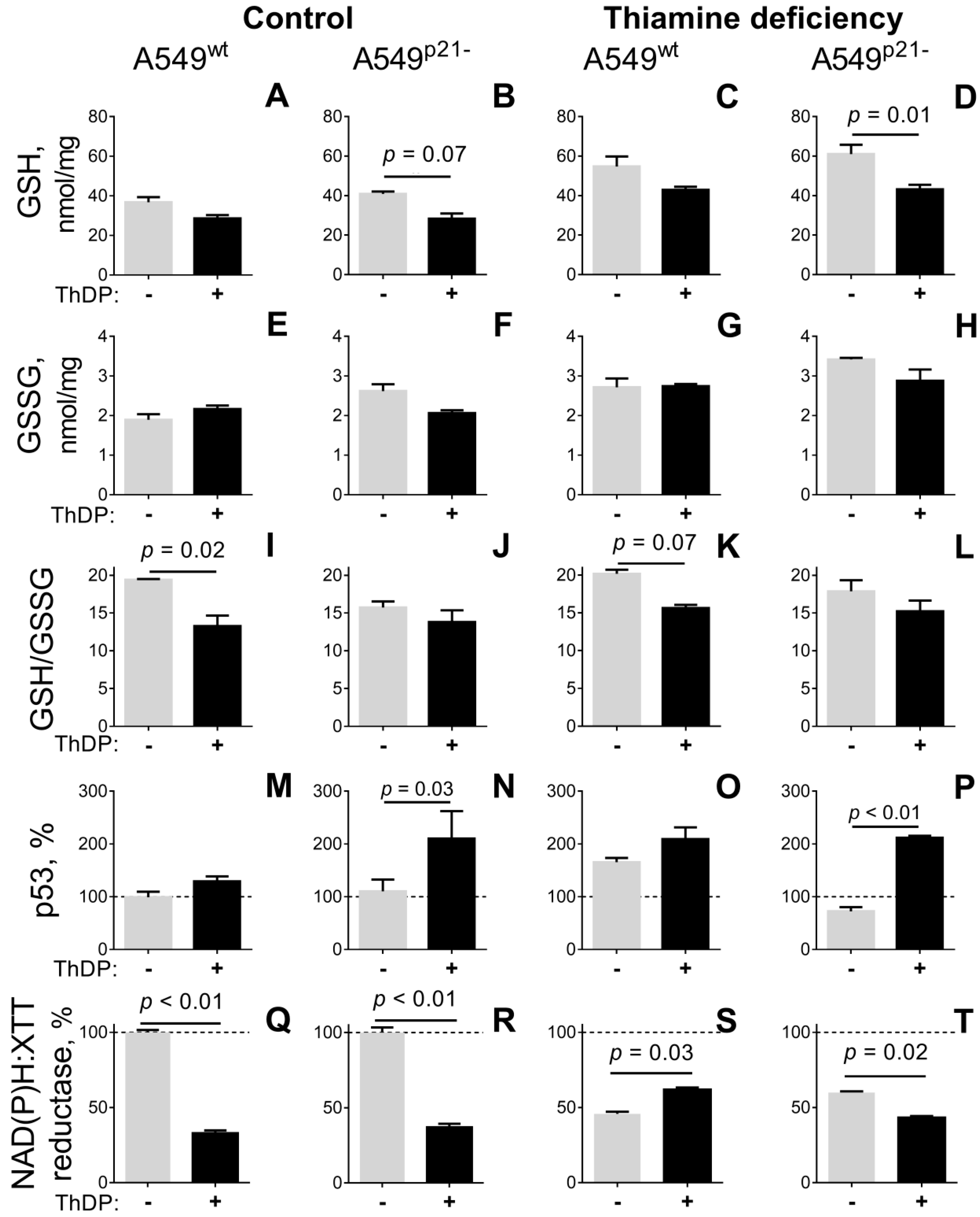
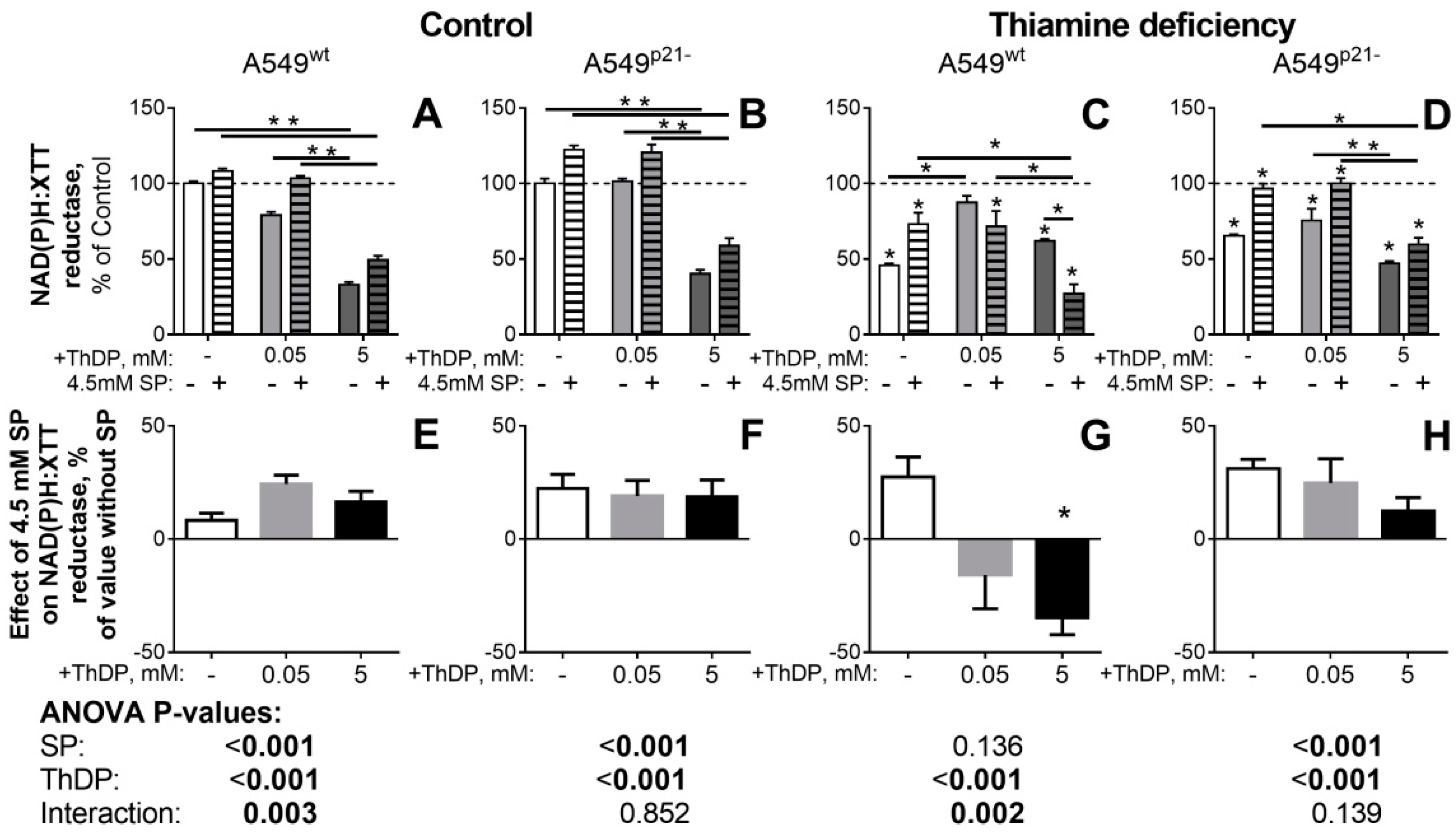
2.3. ThDP Influences Cellular Redox Indicators of A549 Cells in a p5-3 and p21-Dependent Manner
2.4. Inhibitor of OGDHC, Succinyl Phosphonate, Interferes with the Changes in Cellular Redox State, Induced by the OGDHC Activator ThDP
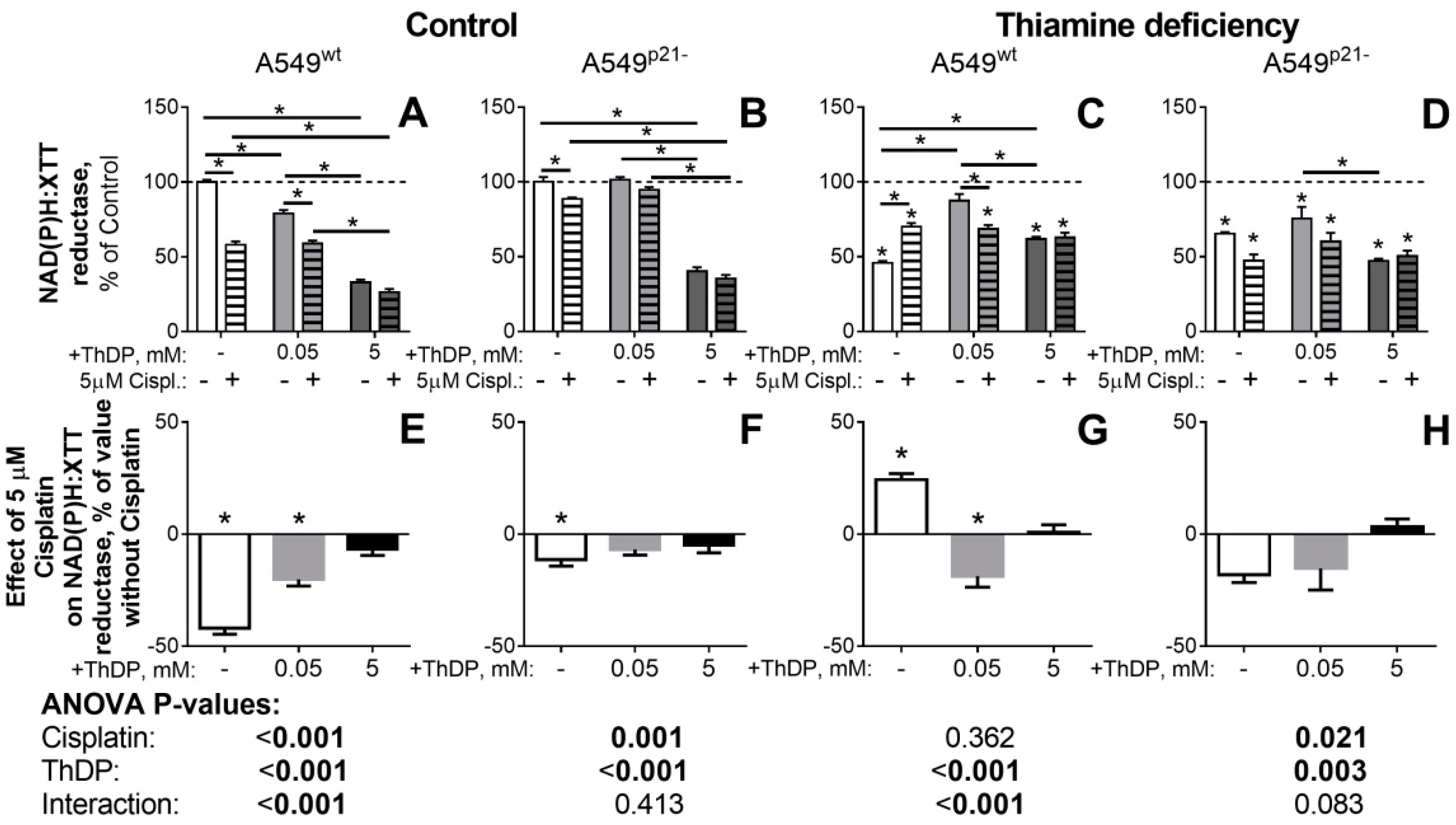
2.5. The p21-Dependent Anticancer Drug Cisplatin Has Similar and Non-Additive to Thiamine/ThDP Actions on Viability of A549 Cells
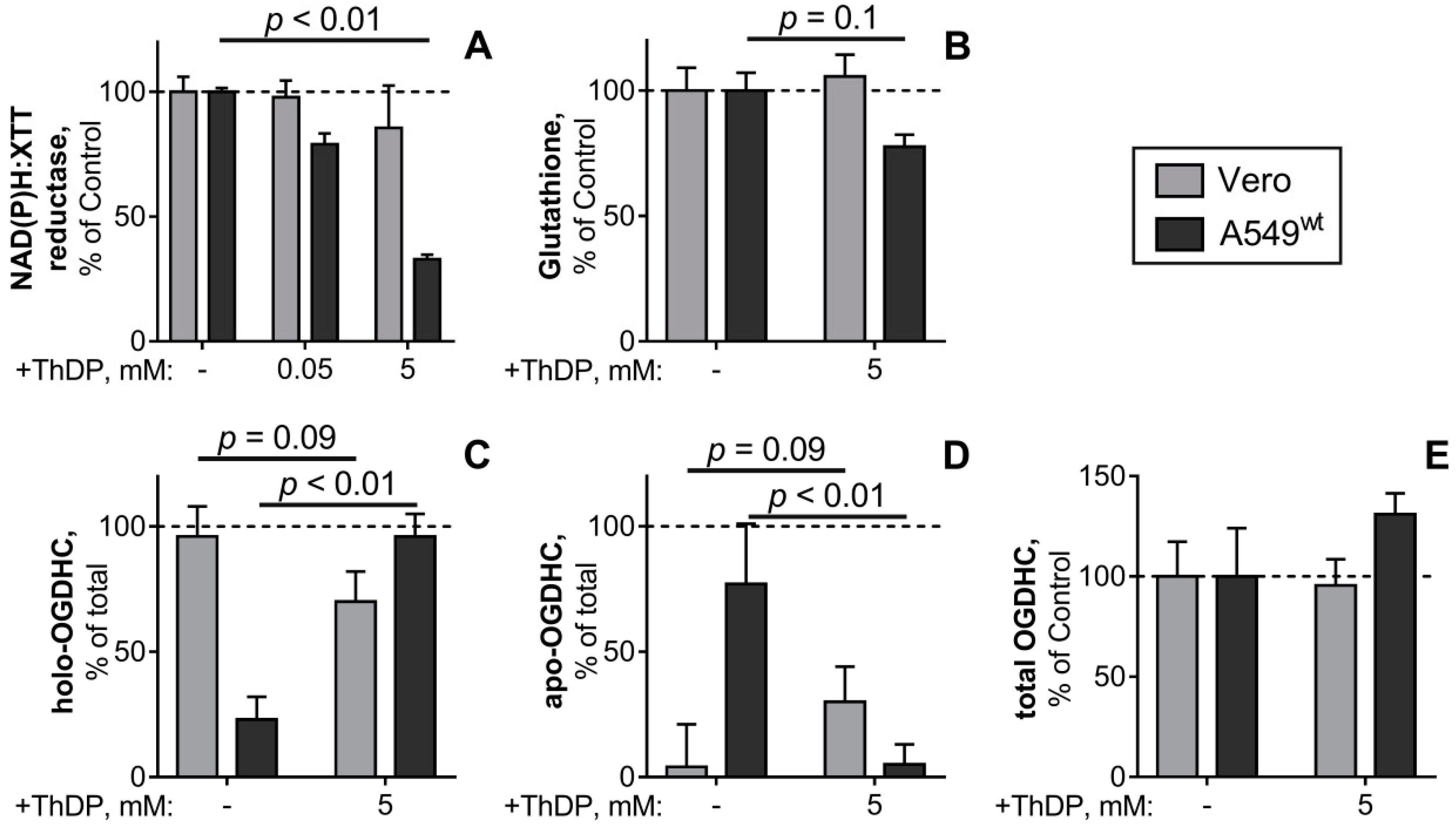
2.6. Comparison of the ThDP Action in the Non-Cancer and Cancer Epihtelial Cell Lines
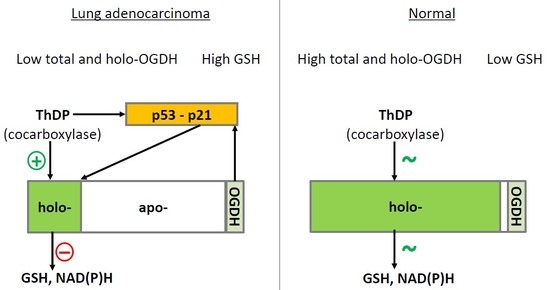
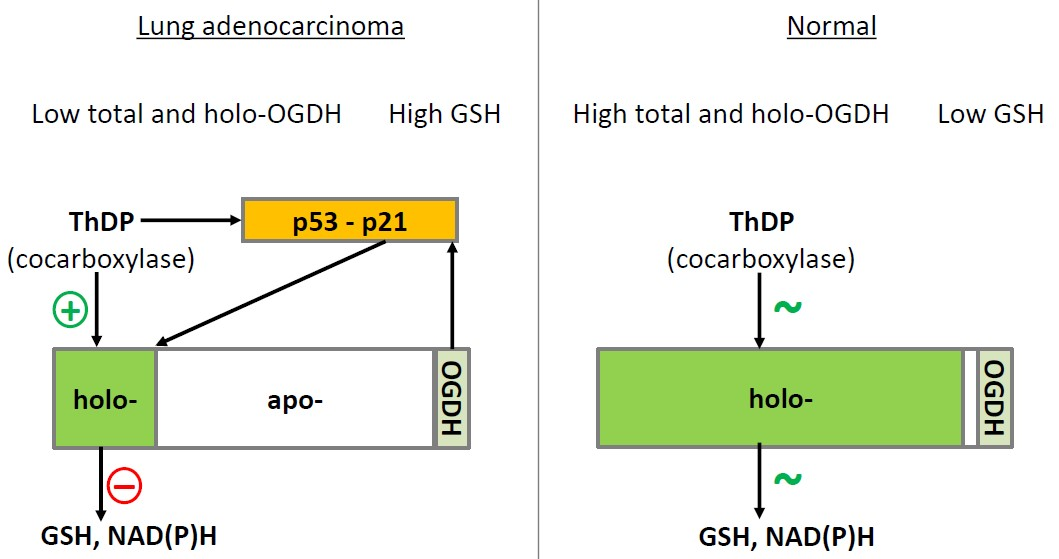
3. Discussion
4. Materials and Methods
4.1. Cell Cultivation and Reagents
4.2. Cellular Model of Thiamine Deficiency
4.3. Construction of Stable p21 Knockdown A549 Cell Line
4.4. Cell Viability Assay
4.5. Preparation of Cell Lysates
4.6. OGDHC Activity Assays
4.7. Assay of p53 by Western Blotting
4.8. Preparation of Methanol–Acetic Acid Extracts
4.9. Glutathione Assay
4.10. Total Protein Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OGDHC | 2-oxoglutarate dehydrogenase complex |
| ROS | reactive oxygen species |
| ThDP | thiamine diphosphate, or cocarboxylase |
| ThMP | thiamine monophosphate |
| SP | succinyl phosphonate |
| XTT | 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide |
References
- Bunik, V. Vitamin-Dependent Multienzyme Complexes of 2-Oxo Acid Dehydrogenases: Structure, Function, Regulation and Medical Implications; Nova Science Publishers: New York, NY, USA, 2017; pp. 1–197. [Google Scholar]
- Bunik, V.I.; Fernie, A.R. Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction: A cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem. J. 2009, 422, 405–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunik, V. Redox-Driven Signaling: 2-Oxo Acid Dehydrogenase Complexes as Sensors and Transmitters of Metabolic Imbalance. Antioxid. Redox Signal. 2019, 30, 1911–1947. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Artiukhov, A.; Kazantsev, A.; Goncalves, R.; Daloso, D.; Oppermann, H.; Kulakovskaya, E.; Lukashev, N.; Fernie, A.; Brand, M.; et al. Specific inhibition by synthetic analogs of pyruvate reveals that the pyruvate dehydrogenase reaction is essential for metabolism and viability of glioblastoma cells. Oncotarget 2015, 6, 40036–40052. [Google Scholar] [CrossRef]
- Bunik, V.I.; Mkrtchyan, G.; Grabarska, A.; Oppermann, H.; Daloso, D.; Araujo, W.L.; Juszczak, M.; Rzeski, W.; Bettendorff, L.; Fernie, A.R.; et al. Inhibition of mitochondrial 2-oxoglutarate dehydrogenase impairs viability of cancer cells in a cell-specific metabolism-dependent manner. Oncotarget 2016, 7, 26400–26421. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.L.; Ulanet, D.B.; Pirman, D.; Mahoney, C.E.; Coco, J.; Si, Y.; Chen, Y.; Huang, L.; Ren, J.; Choe, S.; et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep. 2016, 17, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Li, D.; Peng, H.L.; Laroche, F.J.; Mansour, M.R.; Gjini, E.; Aioub, M.; Helman, D.J.; Roderick, J.E.; Cheng, T.; et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia 2016, 30, 1365–1374. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.O.; Kim, M.S.; Ostrow, K.L.; Liu, J.; Wisman, G.B.; Park, H.L.; Poeta, M.L.; Jeronimo, C.; Henrique, R.; Lendvai, A.; et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008, 68, 2661–2670. [Google Scholar] [CrossRef] [Green Version]
- Ostrow, K.L.; Park, H.L.; Hoque, M.O.; Kim, M.S.; Liu, J.; Argani, P.; Westra, W.; Van Criekinge, W.; Sidransky, D. Pharmacologic unmasking of epigenetically silenced genes in breast cancer. Clin. Cancer Res. 2009, 15, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, M.S.; Kudryavtseva, A.V.; Lakunina, V.A.; Snezhkina, A.V.; Volchenko, N.N.; Slavnova, E.N.; Danilova, T.V.; Sadritdinova, A.F.; Melnikova, N.V.; Belova, A.A.; et al. Downregulation of OGDHL expression is associated with promoter hypermethylation in colorectal cancer. Mol. Biol. 2015, 49, 678–688. [Google Scholar] [CrossRef]
- Sen, T.; Sen, N.; Noordhuis, M.G.; Ravi, R.; Wu, T.C.; Ha, P.K.; Sidransky, D.; Hoque, M.O. OGDHL is a modifier of AKT-dependent signaling and NF-kappaB function. PLoS ONE 2012, 7, e48770. [Google Scholar] [CrossRef] [Green Version]
- McLure, K.G.; Takagi, M.; Kastan, M.B. NAD+ modulates p53 DNA binding specificity and function. Mol. Cell. Biol. 2004, 24, 9958–9967. [Google Scholar] [CrossRef] [Green Version]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of the non-coenzyme action of thiamin: Protein targets and medical significance. Biochemistry 2019, 84, 829–850. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.D.; Moscow, J.A. Can an enzyme cofactor be a factor in malignant progression? Cancer Biol. Ther. 2010, 10, 1112–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boros, L.G. Population thiamine status and varying cancer rates between western, Asian and African countries. Anticancer Res. 2000, 20, 2245–2248. [Google Scholar] [PubMed]
- Duan, L.; Perez, R.E.; Chen, L.; Blatter, L.A.; Maki, C.G. p53 promotes AKT and SP1-dependent metabolism through the pentose phosphate pathway that inhibits apoptosis in response to Nutlin-3a. J. Mol. Cell Biol. 2018, 10, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Daily, A.; Liu, S.; Bhatnagar, S.; Karabakhtsian, R.G.; Moscow, J.A. Low-thiamine diet increases mammary tumor latency in FVB/N-Tg(MMTVneu) mice. Int. J. Vitam. Nutr. Res. 2012, 82, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Huang, H.; Lu, X.; Golinski, M.; Comesse, S.; Watt, D.; Grossman, R.B.; Moscow, J.A. Down-regulation of thiamine transporter THTR2 gene expression in breast cancer and its association with resistance to apoptosis. Mol. Cancer Res. MCR 2003, 1, 665–673. [Google Scholar]
- Liu, X.; Lam, E.K.; Wang, X.; Zhang, J.; Cheng, Y.Y.; Lam, Y.W.; Ng, E.K.; Yu, J.; Chan, F.K.; Jin, H.; et al. Promoter hypermethylation mediates downregulation of thiamine receptor SLC19A3 in gastric cancer. Tumour Biol. 2009, 30, 242–248. [Google Scholar] [CrossRef]
- Sweet, R.; Paul, A.; Zastre, J. Hypoxia induced upregulation and function of the thiamine transporter, SLC19A3 in a breast cancer cell line. Cancer Biol. Ther. 2010, 10, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Tsai, H.K.; Lehrer, J.; Alshalalfa, M.; Erho, N.; Davicioni, E.; Lotan, T.L. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer 2017, 17, 759. [Google Scholar] [CrossRef] [Green Version]
- Sikora, M.J.; Cooper, K.L.; Bahreini, A.; Luthra, S.; Wang, G.; Chandran, U.R.; Davidson, N.E.; Dabbs, D.J.; Welm, A.L.; Oesterreich, S. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014, 74, 1463–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattie, M.; Raitano, A.; Morrison, K.; Morrison, K.; An, Z.; Capo, L.; Verlinsky, A.; Leavitt, M.; Ou, J.; Nadell, R.; et al. The Discovery and Preclinical Development of ASG-5ME, an Antibody-Drug Conjugate Targeting SLC44A4-Positive Epithelial Tumors Including Pancreatic and Prostate Cancer. Mol. Cancer Ther. 2016, 15, 2679–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.C.; Whetstine, J.R.; Witt, T.L.; Schuetz, J.D.; Matherly, L.H. Repression of human reduced folate carrier gene expression by wild type p53. J. Biol. Chem. 2001, 276, 8713–8719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytovchenko, O.; Kunji, E.R.S. Expression and putative role of mitochondrial transport proteins in cancer. Biochim. Biophys. Acta. Bioenerg. 2017, 1858, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Burch, T.C.; Rhim, J.S.; Nyalwidhe, J.O. Mitochondria Biogenesis and Bioenergetics Gene Profiles in Isogenic Prostate Cells with Different Malignant Phenotypes. Biomed Res. Int. 2016, 2016, 1785201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sertel, S.; Eichhorn, T.; Sieber, S.; Sauer, A.; Weiss, J.; Plinkert, P.K.; Efferth, T. Factors determining sensitivity or resistance of tumor cell lines towards artesunate. Chem. Biol. Interact. 2010, 185, 42–52. [Google Scholar] [CrossRef]
- Jonus, H.C.; Hanberry, B.S.; Khatu, S.; Kim, J.; Luesch, H.; Dang, L.H.; Bartlett, M.G.; Zastre, J.A. The adaptive regulation of thiamine pyrophosphokinase-1 facilitates malignant growth during supplemental thiamine conditions. Oncotarget 2018, 9, 35422–35438. [Google Scholar] [CrossRef]
- Lu’o’ng, K.V.; Nguyen, L.T. The role of thiamine in cancer: Possible genetic and cellular signaling mechanisms. Cancer Genom. Proteom. 2013, 10, 169–185. [Google Scholar]
- Comin-Anduix, B.; Boren, J.; Martinez, S.; Moro, C.; Centelles, J.J.; Trebukhina, R.; Petushok, N.; Lee, W.N.; Boros, L.G.; Cascante, M. The effect of thiamine supplementation on tumour proliferation. A metabolic control analysis study. Eur. J. Biochem. 2001, 268, 4177–4182. [Google Scholar] [CrossRef]
- Hanberry, B.S.; Berger, R.; Zastre, J.A. High-dose vitamin B1 reduces proliferation in cancer cell lines analogous to dichloroacetate. Cancer Chemother. Pharmacol. 2014, 73, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Montissol, S.; Uber, A.; Ganley, S.; Grossestreuer, A.V.; Berg, K.; Heydrick, S.; Donnino, M.W. The Effects of Thiamine on Breast Cancer Cells. Molecules 2018, 23, 1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyko, A.; Ksenofontov, A.; Ryabov, S.; Baratova, L.; Graf, A.; Bunik, V. Delayed Influence of Spinal Cord Injury on the Amino Acids of NO(*) Metabolism in Rat Cerebral Cortex Is Attenuated by Thiamine. Front. Med. 2017, 4, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schupp, N.; Dette, E.M.; Schmid, U.; Bahner, U.; Winkler, M.; Heidland, A.; Stopper, H. Benfotiamine reduces genomic damage in peripheral lymphocytes of hemodialysis patients. Naunyn-Schmiedebergs Arch. Pharmacol. 2008, 378, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (thiamine) and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef]
- Wang, X.; Xu, M.; Frank, J.A.; Ke, Z.J.; Luo, J. Thiamine deficiency induces endoplasmic reticulum stress and oxidative stress in human neurons derived from induced pluripotent stem cells. Toxicol. Appl. Pharmacol. 2017, 320, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Parkhomenko, Y.M.; Kudryavtsev, P.A.; Pylypchuk, S.Y.; Chekhivska, L.I.; Stepanenko, S.P.; Sergiichuk, A.A.; Bunik, V.I. Chronic alcoholism in rats induces a compensatory response, preserving brain thiamine diphosphate, but the brain 2-oxo acid dehydrogenases are inactivated despite unchanged coenzyme levels. J. Neurochem. 2011, 117, 1055–1065. [Google Scholar] [CrossRef]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J., Jr.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef]
- Xu, S.; Huang, H.; Chen, Y.N.; Deng, Y.T.; Zhang, B.; Xiong, X.D.; Yuan, Y.; Zhu, Y.; Huang, H.; Xie, L.; et al. DNA damage responsive miR-33b-3p promoted lung cancer cells survival and cisplatin resistance by targeting p21(WAF1/CIP1). Cell Cycle 2016, 15, 2920–2930. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, B.H.; Nielsen, D.; Thorsteinsdottir, U.A.; Hoffmann, E.K.; Lambert, I.H. Downregulation of LRRC8A protects human ovarian and alveolar carcinoma cells against Cisplatin-induced expression of p53, MDM2, p21Waf1/Cip1, and Caspase-9/-3 activation. Am. J. Physiol. Cell Physiol. 2016, 310, C857–C873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cooper, A.J.; Krasnikov, B.F.; Xu, H.; Bubber, P.; Pinto, J.T.; Gibson, G.E.; Hanigan, M.H. Cisplatin-induced toxicity is associated with platinum deposition in mouse kidney mitochondria in vivo and with selective inactivation of the alpha-ketoglutarate dehydrogenase complex in LLC-PK1 cells. Biochemistry 2006, 45, 8959–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talwar, D.; Davidson, H.; Cooney, J.; St, J.R.D. Vitamin B(1) status assessed by direct measurement of thiamin pyrophosphate in erythrocytes or whole blood by HPLC: Comparison with erythrocyte transketolase activation assay. Clin. Chem. 2000, 46, 704–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunik, V.; Raddatz, G.; Strumilo, S. Translating enzymology into metabolic regulation: The case of the 2-oxoglutarate dehydrogenase multienzyme complex. Curr. Chem. Biol. 2013, 7, 74–93. [Google Scholar] [CrossRef]
- Ksenofontov, A.L.; Boyko, A.I.; Mkrtchyan, G.V.; Tashlitsky, V.N.; Timofeeva, A.V.; Graf, A.V.; Bunik, V.I.; Baratova, L.A. Analysis of Free Amino Acids in Mammalian Brain Extracts. Biochem. Biokhimiia 2017, 82, 1183–1192. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.D. Straightforward Statistics for the Behavioral Sciences; Brooks/Cole Publishing Company: Pacific Grove, CA, USA, 1996. [Google Scholar]
- Zhang, D.G.; Zhang, J.; Mao, L.L.; Wu, J.X.; Cao, W.J.; Zheng, J.N.; Pei, D.S. p21-Activated kinase 5 affects cisplatin-induced apoptosis and proliferation in hepatocellular carcinoma cells. Tumour Biol. 2015, 36, 3685–3691. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Q.; Yang, X.; Zhao, M.; Yang, T.; Yao, A.; Tian, X. PACT cessation overcomes ovarian cancer cell chemoresistance to cisplatin by enhancing p53-mediated apoptotic pathway. Biochem. Biophys. Res. Commun. 2019, 511, 719–724. [Google Scholar] [CrossRef]
- Guo, Q.; Lan, F.; Yan, X.; Xiao, Z.; Wu, Y.; Zhang, Q. Hypoxia exposure induced cisplatin resistance partially via activating p53 and hypoxia inducible factor-1alpha in non-small cell lung cancer A549 cells. Oncol. Lett. 2018, 16, 801–808. [Google Scholar] [CrossRef]
- Sappington, D.R.; Siegel, E.R.; Hiatt, G.; Desai, A.; Penney, R.B.; Jamshidi-Parsian, A.; Griffin, R.J.; Boysen, G. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim. Biophys. Acta 2016, 1860, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; DeGraff, W.; Friedman, N.; Mitchell, J.B. Selective modulation of glutathione levels in human normal versus tumor cells and subsequent differential response to chemotherapy drugs. Cancer Res. 1986, 46, 2845–2848. [Google Scholar]
- Markovic, J.; Garcia-Gimenez, J.L.; Gimeno, A.; Vina, J.; Pallardo, F.V. Role of glutathione in cell nucleus. Free Radic. Res. 2010, 44, 721–733. [Google Scholar] [CrossRef]
- Walther, U.I.; Muckter, H.; Fichtl, B.; Forth, W. Influence of glutathione on zinc-mediated cellular toxicity. Biol. Trace Elem. Res. 1999, 67, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.L.; Mitchell, J.B.; de Graff, W.; Russo, A.; Glatstein, E. Variation in sensitizing efficiency for SR 2508 in human cells dependent on glutathione content. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1627–1635. [Google Scholar] [CrossRef]
- Rahman, I.; Li, X.Y.; Donaldson, K.; Harrison, D.J.; MacNee, W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am. J. Physiol. 1995, 269, L285–L292. [Google Scholar] [CrossRef] [PubMed]
- Gori, S.S.; Lorkiewicz, P.; Ehringer, D.S.; Belshoff, A.C.; Higashi, R.M.; Fan, T.W.; Nantz, M.H. Profiling thiol metabolites and quantification of cellular glutathione using FT-ICR-MS spectrometry. Anal. Bioanal. Chem. 2014, 406, 4371–4379. [Google Scholar] [CrossRef]
- Rao, M.; Kumar, M.M.; Rao, M.A. In vitro and in vivo effects of phenolic antioxidants against cisplatin-induced nephrotoxicity. J. Biochem. 1999, 125, 383–390. [Google Scholar] [CrossRef]
- Cao, Z.; Ding, Y.; Cao, L.; Ding, G.; Wang, Z.; Xiao, W. Isochlorogenic acid C prevents enterovirus 71 infection via modulating redox homeostasis of glutathione. Sci. Rep. 2017, 7, 16278. [Google Scholar] [CrossRef] [Green Version]
- Okai, Y.; Higashi-Okai, K.; Sato, E.F.; Konaka, R.; Inoue, M. Potent radical-scavenging activities of thiamin and thiamin diphosphate. J. Clin. Biochem. Nutr. 2007, 40, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Stepuro, I.I.; Oparin, A.Y.; Stsiapura, V.I.; Maskevich, S.A.; Titov, V.Y. Oxidation of thiamine on reaction with nitrogen dioxide generated by ferric myoglobin and hemoglobin in the presence of nitrite and hydrogen peroxide. Biochem. Biokhimiia 2012, 77, 41–55. [Google Scholar] [CrossRef]
- Nabokina, S.M.; Inoue, K.; Subramanian, V.S.; Valle, J.E.; Yuasa, H.; Said, H.M. Molecular identification and functional characterization of the human colonic thiamine pyrophosphate transporter. J. Biol. Chem. 2014, 289, 4405–4416. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Chen, J.; Gan, S.; Guan, H.; Zhou, Y.; Ouyang, Q.; Shi, J. DNA damage strength modulates a bimodal switch of p53 dynamics for cell-fate control. BMC Biol. 2013, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Huang, B.; Zhu, Y.; Li, Y.; Liu, F.; Shi, J. Cell type-dependent bimodal p53 activation engenders a dynamic mechanism of chemoresistance. Sci. Adv. 2018, 4, eaat5077. [Google Scholar] [CrossRef] [Green Version]
- Borras, C.; Gomez-Cabrera, M.C.; Vina, J. The dual role of p53: DNA protection and antioxidant. Free Radic. Res. 2011, 45, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Shen, Z.; Wu, D.; Xie, X.; Xu, X.; Lv, L.; Dai, H.; Chen, J.; Gan, X. Glutathione Peroxidase 1 Promotes NSCLC Resistance to Cisplatin via ROS-Induced Activation of PI3K/AKT Pathway. Biomed Res. Int. 2019, 2019, 7640547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Bermudez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; Garcia-Grande, A.; Coronado, M.J.; Laine-Menendez, S.; Palacios-Zambrano, S.; Moreno-Villa, M.R.; Ruiz-Valdepenas, A.M.; Lendinez, C.; et al. Cisplatin resistance involves a metabolic reprogramming through ROS and PGC-1alpha in NSCLC which can be overcome by OXPHOS inhibition. Free Radic. Biol. Med. 2019, 135, 167–181. [Google Scholar] [CrossRef]
- Bunik, V.I.; Brand, M.D. Generation of superoxide and hydrogen peroxide by side reactions of mitochondrial 2-oxoacid dehydrogenase complexes in isolation and in cells. Biol. Chem. 2018, 399, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchyan, G.V.; Graf, A.; Trofimova, L.; Ksenofontov, A.; Baratova, L.; Bunik, V. Positive correlation between rat brain glutamate concentrations and mitochondrial 2-oxoglutarate dehydrogenase activity. Anal. Biochem. 2018, 552, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 12671. [Google Scholar] [CrossRef]
- Allen, E.M.; Mieyal, J.J. Protein-thiol oxidation and cell death: Regulatory role of glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Bayir, H.; Anthonymuthu, T.S.; Tyurina, Y.Y.; Patel, S.J.; Amoscato, A.A.; Lamade, A.M.; Yang, Q.; Vladimirov, G.K.; Philpott, C.C.; Kagan, V.E. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem. Biol. 2020, 27, 387–408. [Google Scholar] [CrossRef]
- Bunik, V.I.; Sievers, C. Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur. J. Biochem. 2002, 269, 5004–5015. [Google Scholar] [CrossRef]
- Bunik, V.; Kaehne, T.; Degtyarev, D.; Shcherbakova, T.; Reiser, G. Novel isoenzyme of 2-oxoglutarate dehydrogenase is identified in brain, but not in heart. FEBS J. 2008, 275, 4990–5006. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Denton, T.T.; Xu, H.; Thompson, C.M.; Cooper, A.J.; Gibson, G.E. Phosphonate analogues of alpha-ketoglutarate inhibit the activity of the alpha-ketoglutarate dehydrogenase complex isolated from brain and in cultured cells. Biochemistry 2005, 44, 10552–10561. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, V.A.; Artiukhov, A.V.; Oppermann, H.; Kazantsev, A.V.; Lukashev, N.V.; Bunik, V.I. Mitochondrial Impairment May Increase Cellular NAD(P)H: Resazurin Oxidoreductase Activity, Perturbing the NAD(P)H-Based Viability Assays. Cells 2015, 4, 427–451. [Google Scholar] [CrossRef] [Green Version]
- Mkrtchyan, G.; Graf, A.; Bettendorff, L.; Bunik, V. Cellular thiamine status is coupled to function of mitochondrial 2-oxoglutarate dehydrogenase. Neurochem. Int. 2016, 101, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Hauke, J.; Kossowski, T. Comparison of Values of Pearson’s and Spearman’s Correlation Coefficients on the Same Sets of Data. Quaest. Geogr. 2011, 30. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Standard DMEM | Thiamine-free DMEM | ||||||
|---|---|---|---|---|---|---|---|---|
| Addition of ThDP for 24h | - | + 5 mM ThDP | - | + 5 mM ThDP | ||||
| Cell Line | A549wt a | A549p21- b | A549wt c | A549p21- d | A549wt e | A549p21- f | A549wt g | A549p21- h |
| Protein, mg | 1.27 ± 0.03 | 1.14 ± 0.07 e,h | 1.08 ± 0.05 e,f,h | 1.13 ± 0.02 e,h | 1.46 ± 0.04 b,c,d,g | 1.38 ± 0.07 c | 1.17 ± 0.05 e,h | 1.44 ± 0.07 b,c,d,g |
| Activity, μmol/(min∙mg) | ||||||||
| total OGDHC∙104 | 3.3 ± 0.8 e,g,h | 5.5 ± 1.4 e,f,g | 4.3 ± 0.3 e,f,g,h | 5.9 ± 0.4 e,f,g | 1.5 ± 0.7 a,b,c,d,g,h | 1.5 ± 0.2 b,c,d,g,h | 8.7 ± 0.2 a,b,c,d,e,f | 8.3 ± 0.3 a,c,e,f |
| apo-OGDHC∙104 | 2.5 ± 0.8 c,e | 3.7 ± 1.4 c,e,f | 0.2 ± 0.3 a,b,g | 2.6 ± 0.4 e,f | 0 ± 0.2 a,b,d,g,h | 0.1 ± 0.2 b,d,g,h | 3.4 ± 0.2 c,e,f | 2.3 ± 0.3 e,f |
| holo-OGDHC∙104 | 0.7 ± 0.3 c,d,g,h | 1.8 ± 0.6 c,g,h | 4.1 ± 0.4 a,b,f | 3.3 ± 0.5 a,h | 2.0 ± 0.3 g,h | 1.4 ± 0.4 c,g,h | 5.3 ± 0.5 a,b,e,f | 6.0 ± 0.5 a,b,d,e,f |
| GSH, nmol/mg | 38 ± 3 e,f | 41 ± 1 f | 29 ± 2 e,f | 28 ± 3 e,f,h | 55 ± 5 a,c,d | 61 ± 5 a,b,c,d,g,h | 43 ± 2 f | 43 ± 2 d,f |
| GSSG, nmol/mg | 1.9 ± 0.1 e,f,g,h | 2.6 ± 0.2 f | 2.2 ± 0.1 f | 2.1 ± 0.1 f,h | 2.8 ± 0.2 a | 3.4 ± 0.1 a,b,c,d | 2.7 ± 0.1 a | 2.9 ± 0.3 a,d |
| GSH/GSSG | 19.4 ± 0.1 c,d | 15.7 ± 0.8 | 13.3 ± 1.4 a,e | 13.8 ± 1.6 a,e | 20.2 ± 0.5 c,d | 17.9 ± 1.5 | 15.6 ± 0.4 | 15.2 ± 1.4 |
| p53, % | 100 ± 10 d,g,h | 110 ± 22 d,g,h | 129 ± 9 | 210 ± 52 a,b,f | 166 ± 8 f | 72 ± 8 d,e,g,h | 209 ± 22 a,b,f | 212 ± 4 a,b,f |
| NAD(P)H:XTT reductase, % | 100 ± 2 c,e,g | 100 ± 3 d,f,h | 33 ± 2 a,g | 40 ± 3 b | 46 ± 1 a | 62 ± 1 b | 65 ± 1 a,c | 47 ± 1 b |
| Cell Line | Vero | A549wt |
|---|---|---|
| GSH, nmol/mg | 1.2 ± 0.1 | 37.7 ± 2.6 |
| Activity, nmol/(min∙mg) | ||
| total OGDHC | 1.76 ± 0.30 | 0.33 ± 0.08 |
| holo-OGDHC | 1.68 ± 0.22 | 0.07 ± 0.03 |
| apo-OGDHC | 0.07 ± 0.30 | 0.25 ± 0.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunik, V.I.; Aleshin, V.A.; Zhou, X.; Tabakov, V.Y.; Karlsson, A. Activation of Mitochondrial 2-Oxoglutarate Dehydrogenase by Cocarboxylase in Human Lung Adenocarcinoma Cells A549 Is p53/p21-Dependent and Impairs Cellular Redox State, Mimicking the Cisplatin Action. Int. J. Mol. Sci. 2020, 21, 3759. https://doi.org/10.3390/ijms21113759
Bunik VI, Aleshin VA, Zhou X, Tabakov VY, Karlsson A. Activation of Mitochondrial 2-Oxoglutarate Dehydrogenase by Cocarboxylase in Human Lung Adenocarcinoma Cells A549 Is p53/p21-Dependent and Impairs Cellular Redox State, Mimicking the Cisplatin Action. International Journal of Molecular Sciences. 2020; 21(11):3759. https://doi.org/10.3390/ijms21113759
Chicago/Turabian StyleBunik, Victoria I., Vasily A. Aleshin, Xiaoshan Zhou, Vyacheslav Yu. Tabakov, and Anna Karlsson. 2020. "Activation of Mitochondrial 2-Oxoglutarate Dehydrogenase by Cocarboxylase in Human Lung Adenocarcinoma Cells A549 Is p53/p21-Dependent and Impairs Cellular Redox State, Mimicking the Cisplatin Action" International Journal of Molecular Sciences 21, no. 11: 3759. https://doi.org/10.3390/ijms21113759
APA StyleBunik, V. I., Aleshin, V. A., Zhou, X., Tabakov, V. Y., & Karlsson, A. (2020). Activation of Mitochondrial 2-Oxoglutarate Dehydrogenase by Cocarboxylase in Human Lung Adenocarcinoma Cells A549 Is p53/p21-Dependent and Impairs Cellular Redox State, Mimicking the Cisplatin Action. International Journal of Molecular Sciences, 21(11), 3759. https://doi.org/10.3390/ijms21113759