Post-Ischemic Renal Fibrosis Progression Is Halted by Delayed Contralateral Nephrectomy: The Involvement of Macrophage Activation

 , ,
, ,  and
and
Abstract
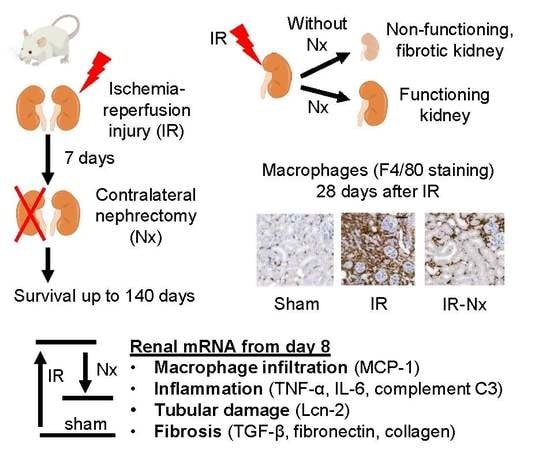
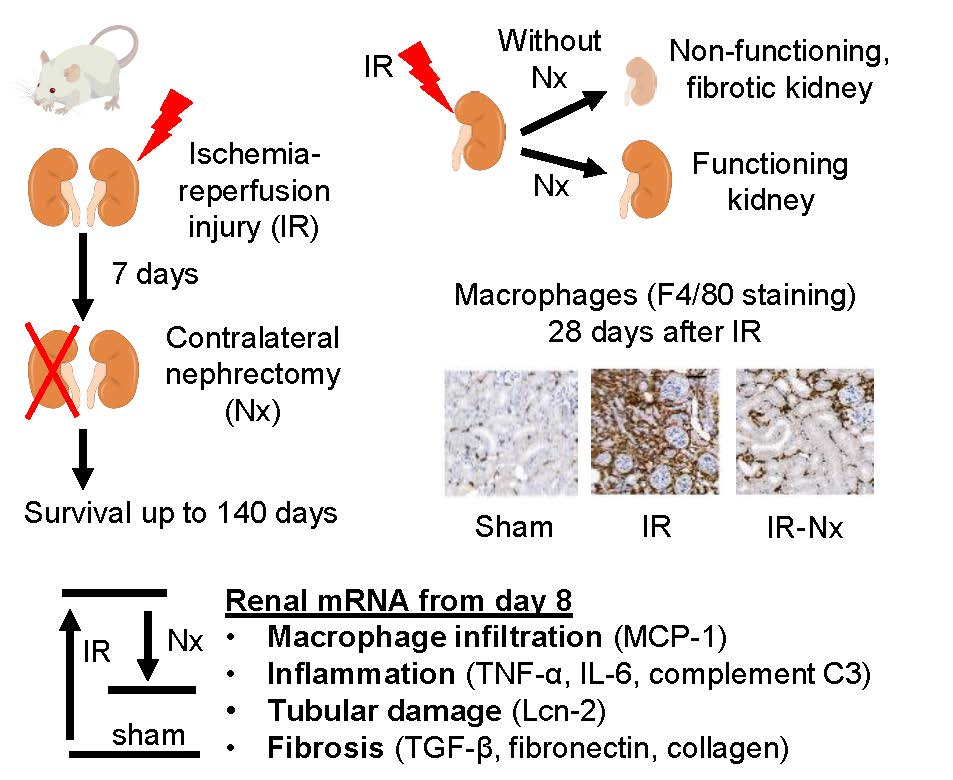
:
1. Introduction
2. Results
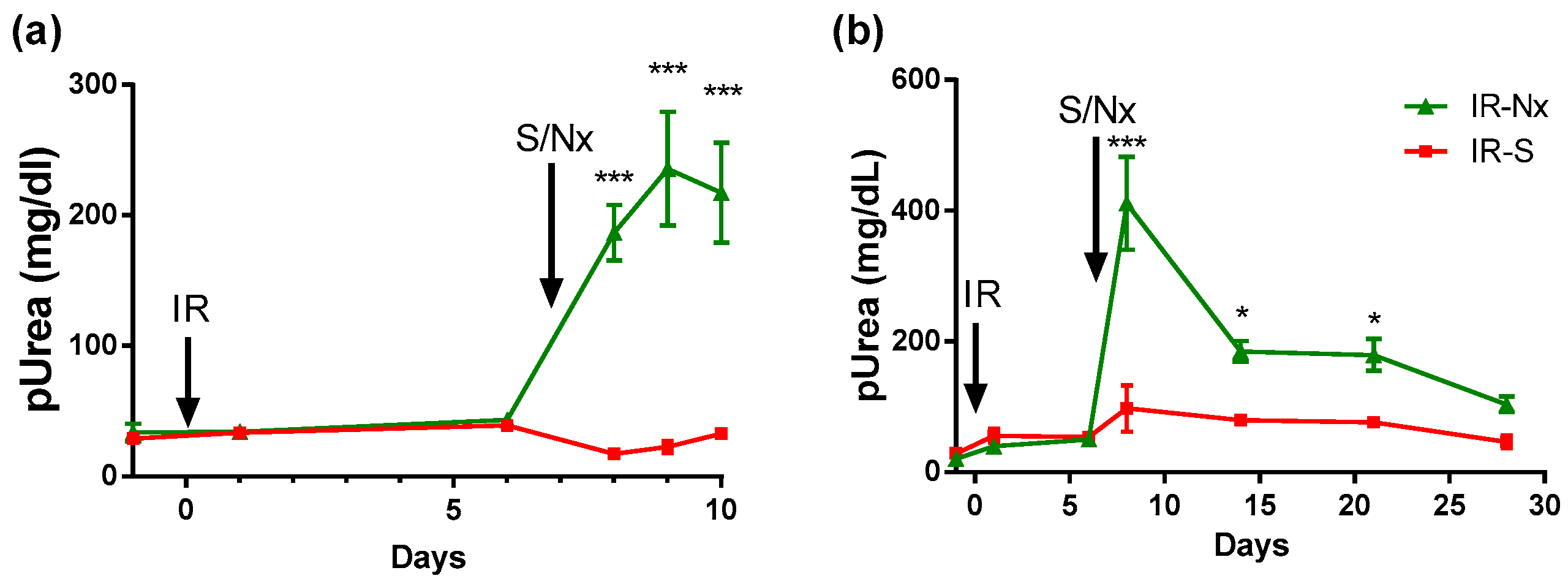
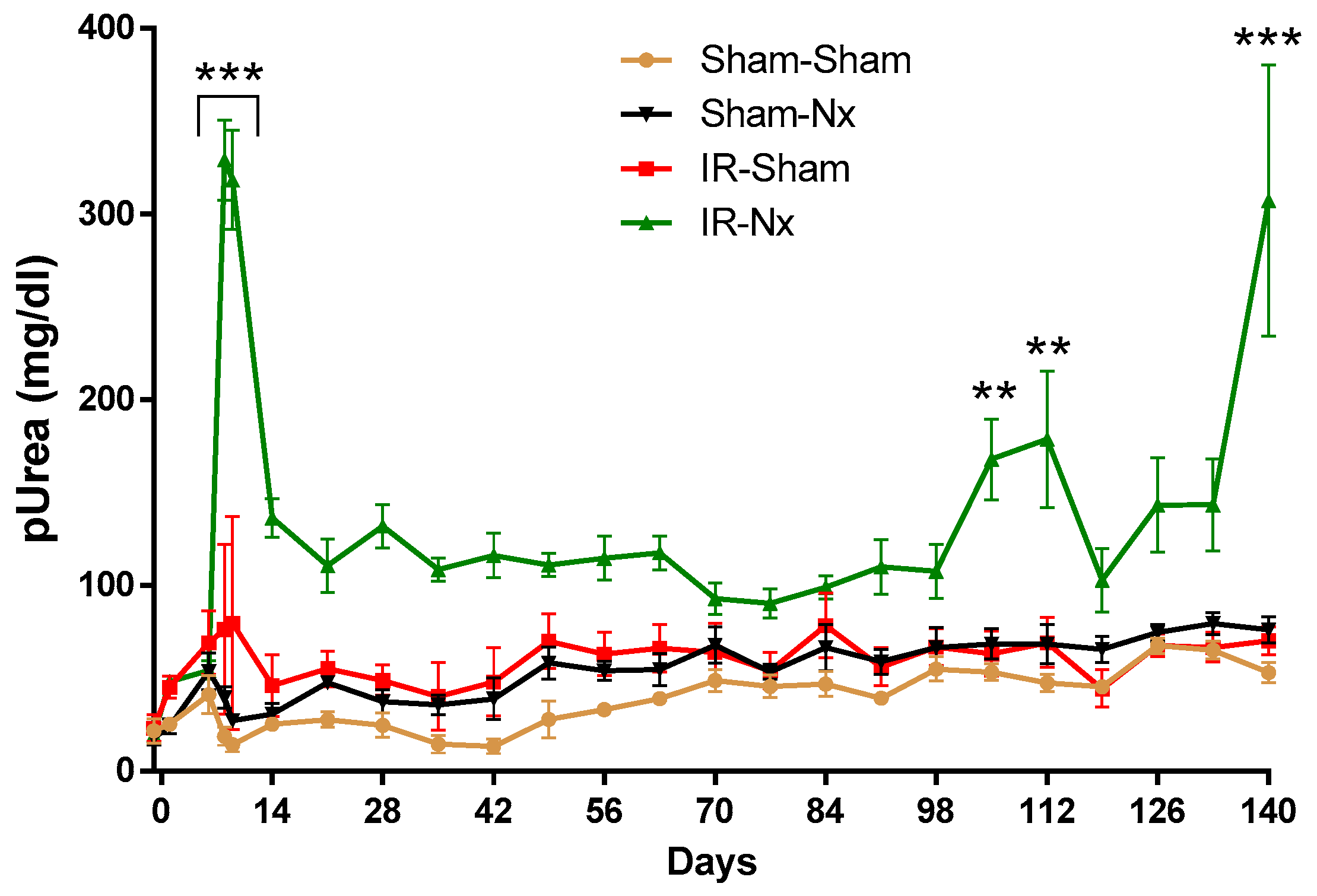
2.1. Delayed Nephrectomy Almost Completely Restored the Function of the Postischemic Kidney
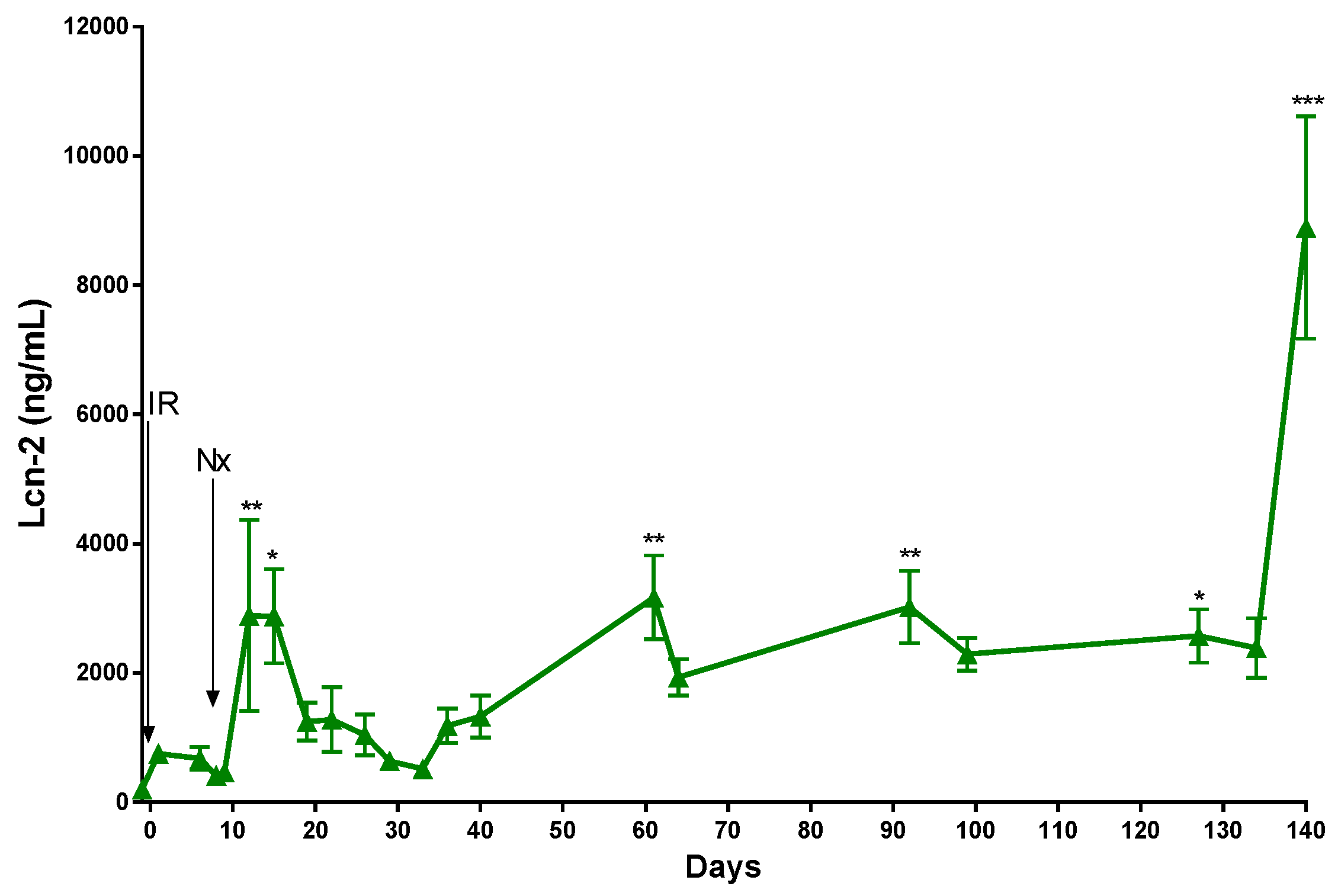
2.2. The Function of the Sole Postischemic Kidney Deteriorated Slowly
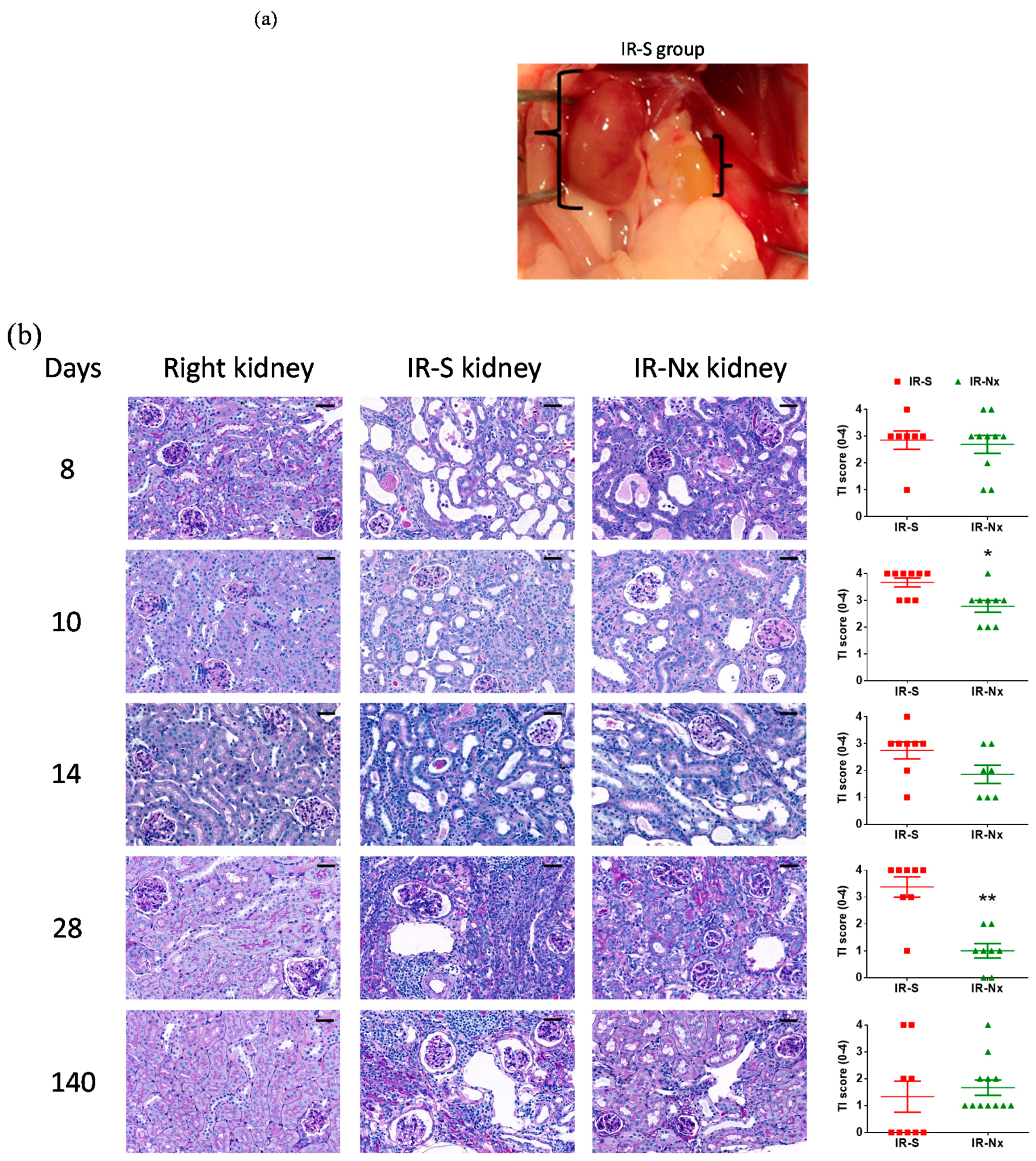
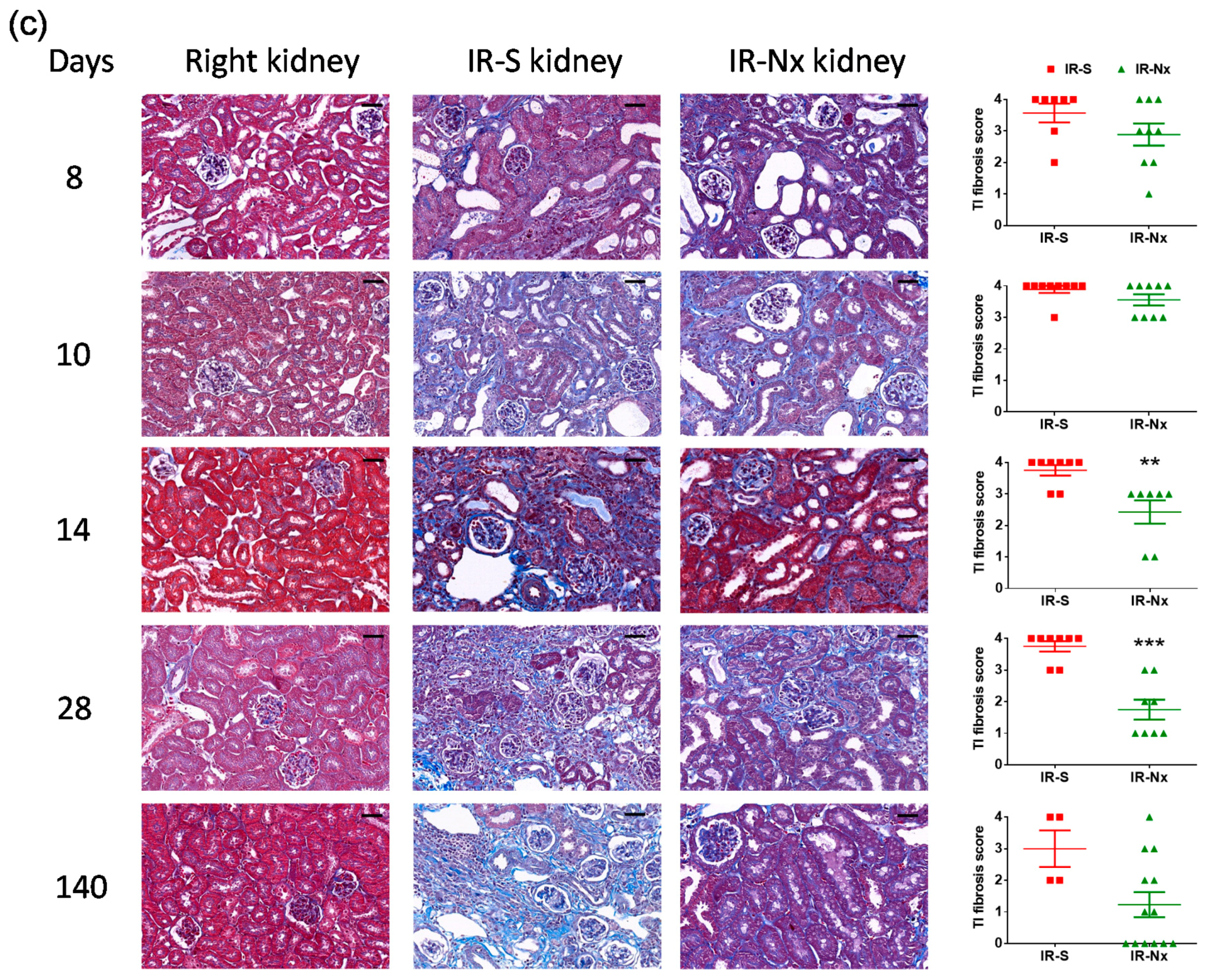
2.3. Histology Demonstrated Partial Regeneration of the Postischemic Kidney after Nx
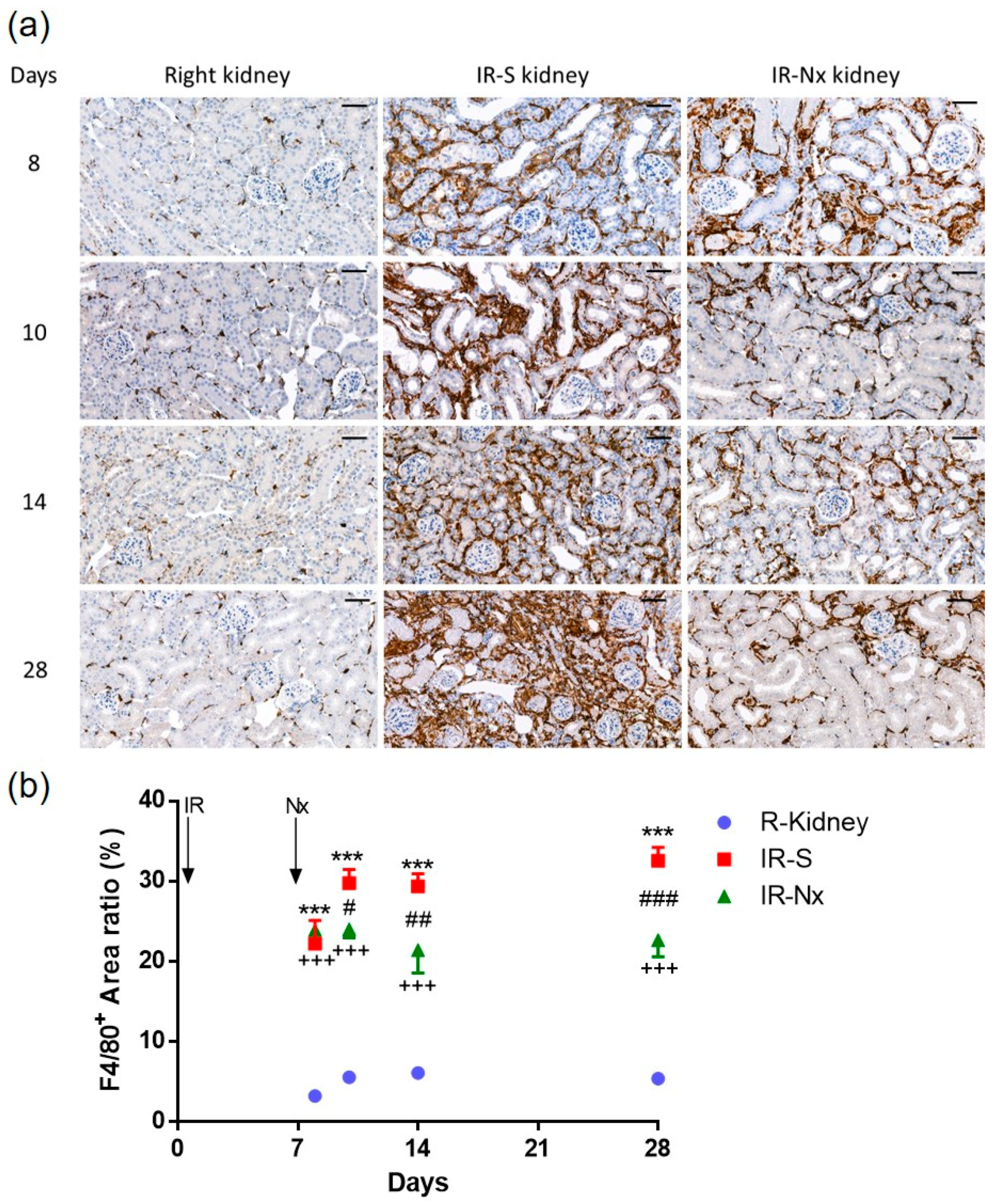
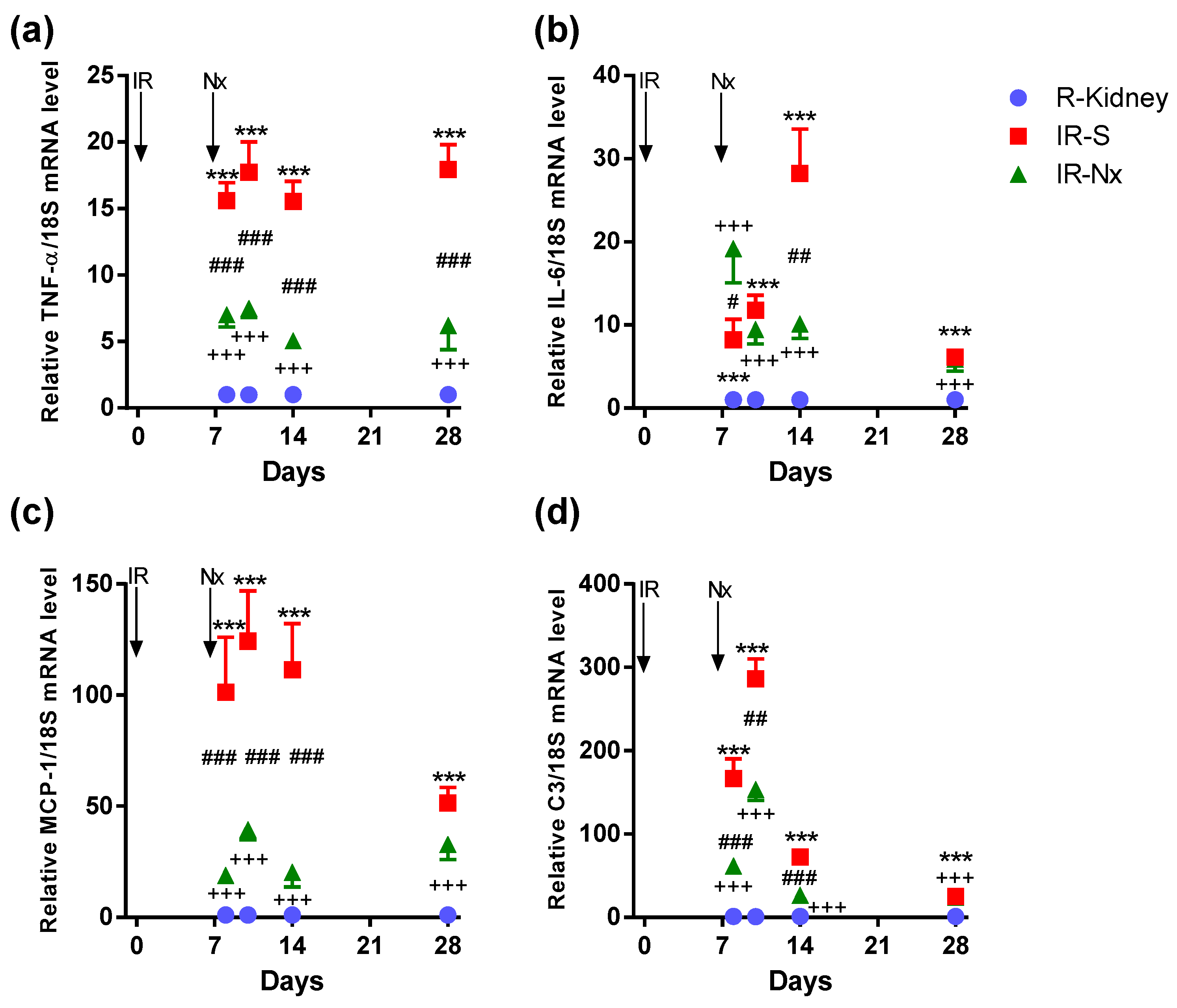
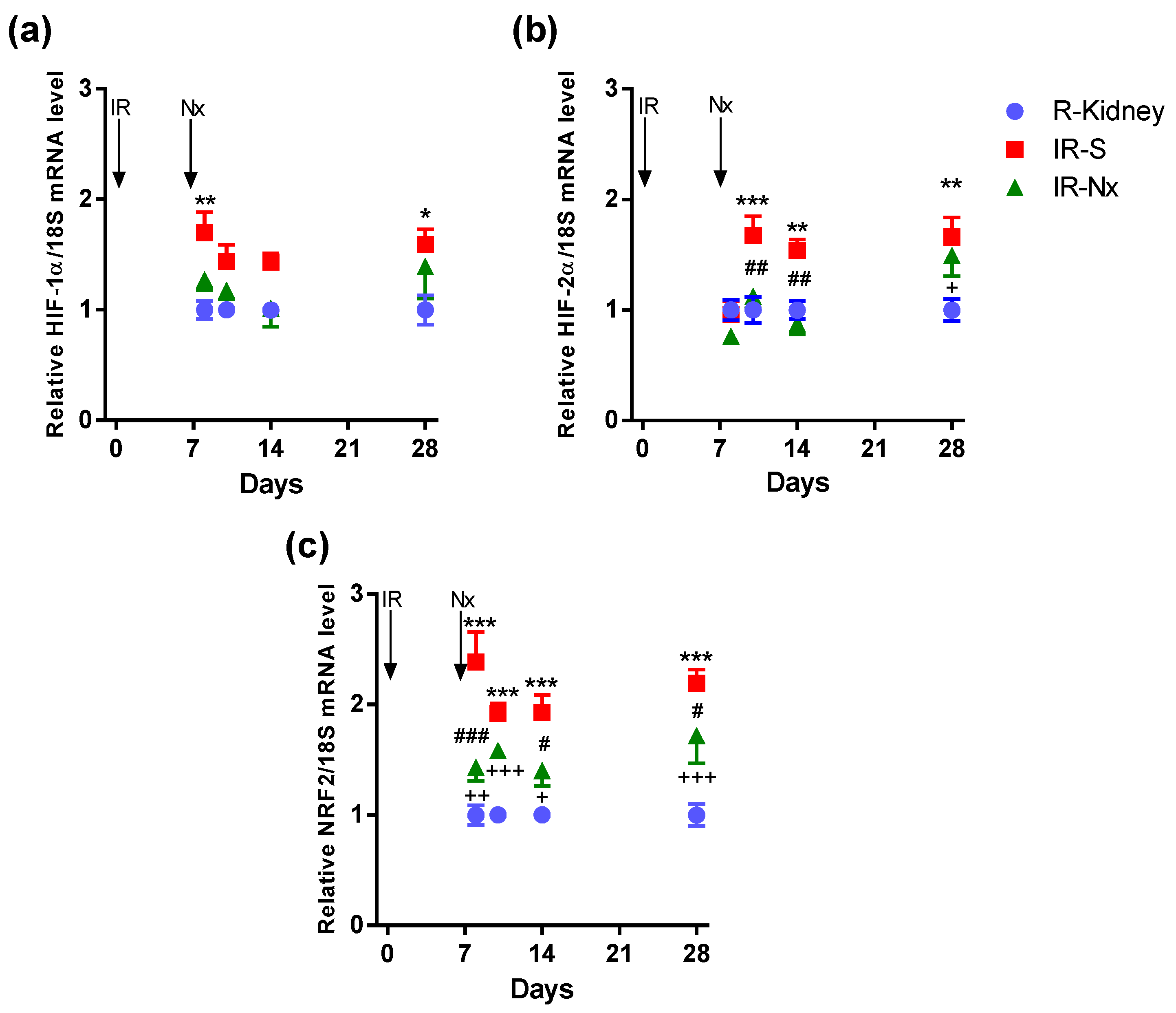
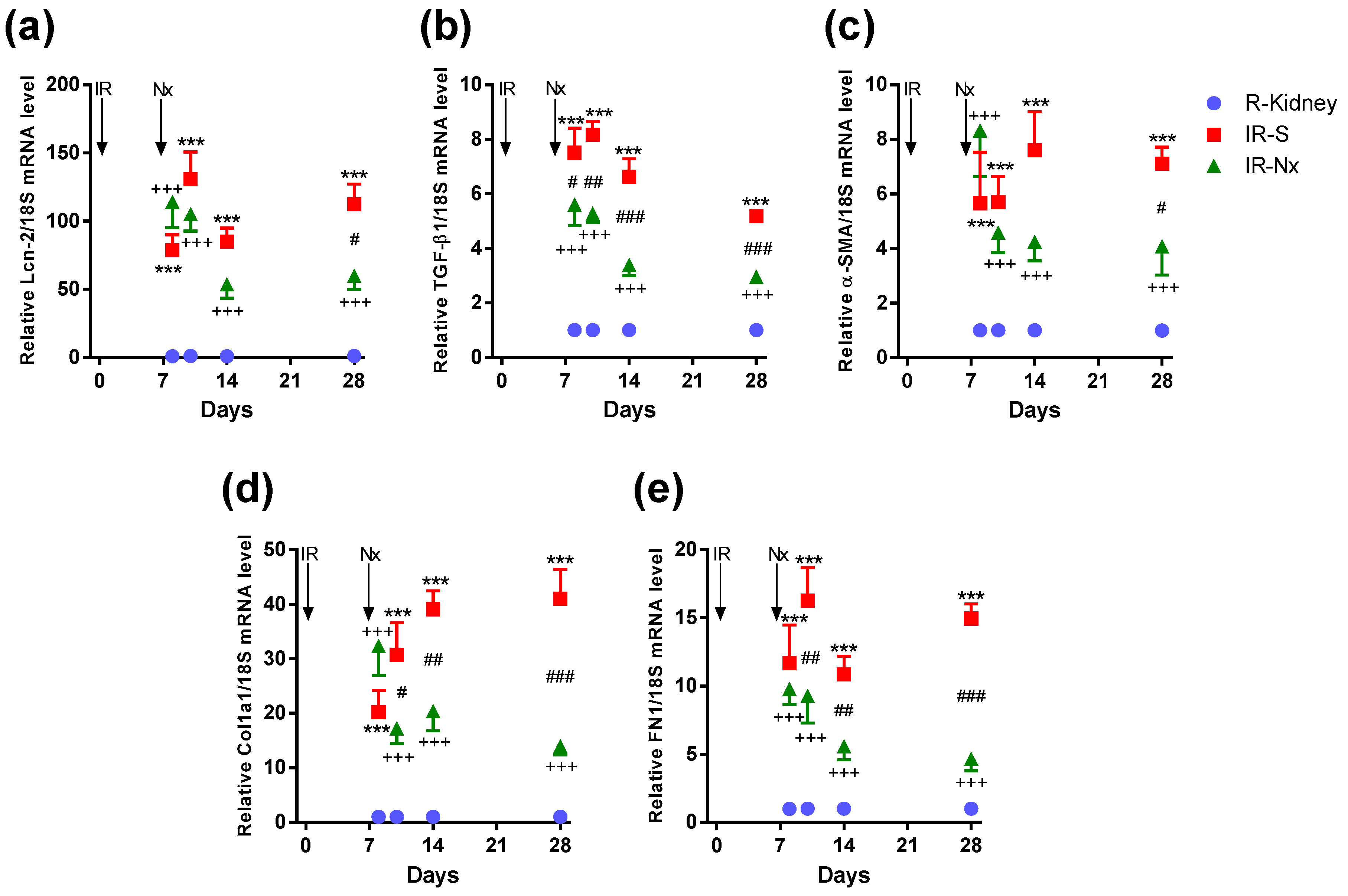
2.4. Nx Downregulated Inflammation-, Hypoxia- and Fibrosis-Related mRNAs in the Postischemic Kidney
3. Discussion
4. Materials and Methods
4.1. Mice
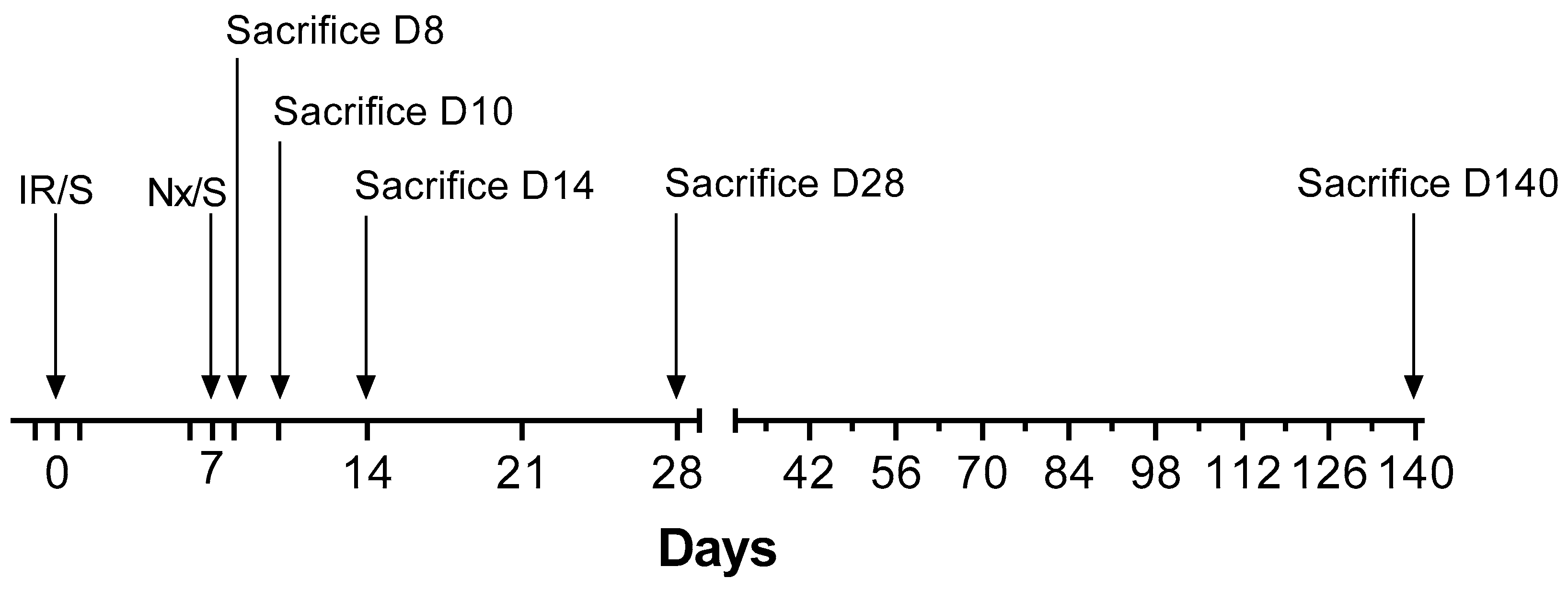
4.2. Experimental Design
4.3. Blood and Urine Sampling and Sacrifice
4.4. Assay of Plasma Urea and Lipocalin-2 (Lcn-2) Concentrations
4.5. Total RNA Isolation and Real Time-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Histology and F4/80 Immunohistochemistry
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kellum, J.A.; Lameire, N.; Aspelin, P.; Barsoum, R.S.; Burdmann, E.A.; Goldstein, S.L.; Herzog, C.A.; Joannidis, M.; Kribben, A.; Levey, A.S.; et al. Kidney disease: Improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int. Suppl. 2012, 2, 1. [Google Scholar]
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortrie, G.; De Geus, H.R.H.; Betjes, M.G.H. The aftermath of acute kidney injury: A narrative review of long-term mortality and renal function. Crit. Care 2019, 23, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Emlet, D.R.; Shaw, A.D.; Kellum, J.A. Sepsis-Associated AKI: Epithelial Cell Dysfunction. Semin. Nephrol. 2015, 35, 85–95. [Google Scholar] [CrossRef]
- Bellomo, R.; Kellum, J.A.; Ronco, C.; Wald, R.; Martensson, J.; Maiden, M.; Bagshaw, S.M.; Glassford, N.J.; Lankadeva, Y.; Vaara, S.T.; et al. Acute kidney injury in sepsis. Intensive Care Med. 2017, 43, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Kanagasundaram, N.S. Pathophysiology of ischaemic acute kidney injury. Ann. Clin. Biochem. 2015, 52, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Sutton, T.A.; Fisher, C.J.; Molitoris, B.A. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002, 62, 1539–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kezić, A.; Stajic, N.; Thaiss, F. Innate immune response in kidney ischemia/reperfusion injury: Potential target for therapy. J. Immunol. Res. 2017, 2017, 6305439. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion-from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet (Lond. Engl.) 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Furuichi, K.; Wada, T.; Iwata, Y.; Kitagawa, K.; Kobayashi, K.I.; Hashimoto, H.; Ishiwata, Y.; Asano, M.; Wang, H.; Matsushima, K.; et al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J. Am. Soc. Nephrol. 2003, 14, 2503–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, S.C.; Cantley, L.G. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr. Nephrol. 2015, 30, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Sharkey, D.; Cantley, L.G. Tubular GM-CSF promotes late MCP-1/CCR2-mediated fibrosis and inflammation after ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2019, 30, 1825–1840. [Google Scholar] [CrossRef]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Tang, P.M.K.; Li, J.; Lan, H.Y. TGF-ß/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Dong, Z. Mouse model of ischemic acute kidney injury: Technical notes and tricks. Am. J. Physiol. Ren. Physiol. 2012, 303. [Google Scholar] [CrossRef] [Green Version]
- Le Clef, N.; Verhulst, A.; D’Haese, P.C.; Vervaet, B.A. Unilateral renal ischemia-reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS ONE 2016, 11, e0152153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Lan, S.; Dieude, M.; Sabo-Vatasescu, J.P.; Karakeussian-Rimbaud, A.; Turgeon, J.; Qi, S.; Gunaratnam, L.; Patey, N.; Hébert, M.J. Caspase-3 Is a Pivotal Regulator of Microvascular Rarefaction and Renal Fibrosis after Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2018, 29, 1900–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, E.E.; Czopek, A.; Clay, M.; Borthwick, G.; Ferenbach, D.; Kluth, D.; Hughes, J. Renal Ischaemia Reperfusion Injury: A Mouse Model of Injury and Regeneration. J. Vis. Exp. 2014, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrypnyk, N.I.; Harris, R.C.; de Caestecker, M.P. Ischemia-reperfusion Model of Acute Kidney Injury and Post Injury Fibrosis in Mice. J. Vis. Exp. 2013, 78, 50496. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int. 2015, 87, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Finn, W.F. Renal counterbalance. J. Lab. Clin. Med. 1985, 105, 523–530. [Google Scholar]
- Kierulf-Lassen, C.; Nielsen, P.M.; Qi, H.; Damgaard, M.; Laustsen, C.; Pedersen, M.; Krag, S.; Birn, H.; Nørregaard, R.; Jespersen, B. Unilateral nephrectomy diminishes ischemic acute kidney injury through enhanced perfusion and reduced pro-inflammatory and pro-fibrotic responses. PLoS ONE 2017, 12, e0190009. [Google Scholar] [CrossRef] [Green Version]
- Polichnowski, A.J.; Griffin, K.A.; Licea-Vargas, H.; Lan, R.; Picken, M.M.; Long, J.; Williamson, G.A.; Rosenberger, C.; Mathia, S.; Venkatachalam, M.A.; et al. Pathophysiology of unilateral ischemia-reperfusion injury: Importance of renal counterbalance and implications for the AKI-CKD transition. Am. J. Physiol. Physiol. 2020, 318, F1086–F1099. [Google Scholar] [CrossRef]
- Khan, M.A.; Assiri, A.M.; Broering, D.C. Complement and macrophage crosstalk during process of angiogenesis in tumor progression. J. Biomed. Sci. 2015, 22, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Odink, K.G.; Fey, G.; Wiebauer, K.; Diggelmann, H. Mouse complement components C3 and C4. Characterization of their messenger RNA and molecular cloning of complementary DNA for C3. J. Biol. Chem. 1981, 256, 1453–1458. [Google Scholar]
- Carroll, M.C. The complement system in regulation of adaptive immunity. Nat. Immunol. 2004, 5, 981–986. [Google Scholar] [CrossRef]
- Goodrum, K.J. Complement component C3 secretion by mouse macrophage-like cell lines. J. Leukoc. Biol. 1987, 41, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Wu, X.; Song, Y.; Chen, Y.; Wan, J. Complement C3 exacerbates renal interstitial fibrosis by facilitating the M1 macrophage phenotype in a mouse model of unilateral ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2019, 317, F1171–F1182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, K.; Liang, X.; Li, Y.; Zhang, Y.; Zhang, C.; Wei, H.; Luo, R.; Ge, S.; Xu, G. Complement C3 produced by macrophages promotes renal fibrosis via IL-17A secretion. Front. Immunol. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, M.; Nematbakhsh, M. Renal ischemia/reperfusion injury; from pathophysiology to treatment. J. Ren. Inj. Prev. 2015, 4, 20–207. [Google Scholar]
- Jang, H.R.; Rabb, H. Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 2015, 11, 88–101. [Google Scholar] [CrossRef]
- Liu, M.; Ning, X.; Li, R.; Yang, Z.; Yang, X.; Sun, S.; Qian, Q. Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell Mol. Med. 2017, 21, 1248–1259. [Google Scholar] [CrossRef]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Tanaka, T.; Nangaku, M. Renal hypoxia in CKD; Pathophysiology and detecting methods. Front. Physiol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Eckardt, K.U.; Bernhardt, W.M.; Weidemann, A.; Warnecke, C.; Rosenberger, C.; Wiesener, M.S.; Willam, C. Role of hypoxia in the pathogenesis of renal disease. Kidney Int. 2005, 68, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Depping, R.; Steinhoff, A.; Schindler, S.G.; Friedrich, B.; Fagerlund, R.; Metzen, E.; Hartmann, E.; Köhler, M. Nuclear translocation of hypoxia-inducible factors (HIFs): Involvement of the classical importin α/β pathway. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 394–404. [Google Scholar] [CrossRef] [Green Version]
- Nezu, M.; Suzuki, N.; Yamamoto, M. Targeting the KEAP1-NRF2 System to Prevent Kidney Disease Progression. Am. J. Nephrol. 2017, 45, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Skrypnyk, N.I.; Voziyan, P.; Yang, H.; de Caestecker, C.R.; Theberge, M.C.; Drouin, M.; Hudson, B.; Harris, R.C.; de Caestecker, M.P. Pyridoxamine reduces postinjury fibrosis and improves functional recovery after acute kidney injury. Am. J. Physiol.—Ren. Physiol. 2016, 311, F268–F277. [Google Scholar] [CrossRef] [PubMed]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chancharoenthana, W.; Leelahavanichkul, A.; Taratummarat, S.; Wongphom, J.; Tiranathanagul, K.; Eiam-Ong, S. Cilostazol attenuates intimal hyperplasia in a mouse model of chronic kidney disease. PLoS ONE 2017, 12, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Kaucsár, T.; Godó, M.; Révész, C.; Kovács, M.; Mócsai, A.; Kiss, N.; Albert, M.; Krenács, T.; Szénási, G.; Hamar, P. Urine/plasma neutrophil gelatinase associated lipocalin ratio is a sensitive and specific marker of subclinical acute kidney injury in mice. PLoS ONE 2016, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Hamar, P.; Song, E.; Kökeny, G.; Chen, A.; Ouyang, N.; Lieberman, J. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2004, 101, 14883–14888. [Google Scholar] [CrossRef] [Green Version]
- Amann, K.; Koch, A.; Hofstetter, J.; Gross, M.L.; Haas, C.; Orth, S.R.; Ehmke, H.; Rump, L.C.; Ritz, E. Glomerulosclerosis and progression: Effect of subantihypertensive doses of α and β blockers. Kidney Int. 2001, 60, 1309–1323. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Duration | Groups | |||
|---|---|---|---|---|
| IR-S | IR-Nx | S-S | S-Nx | |
| 8 days | n = 7 | n = 10 | - | - |
| 10 days | n = 9 | n = 9 | - | - |
| 14 days | n = 8 | n = 7 | - | - |
| 28 days | n = 8 | n = 8 | - | - |
| 140 days | n = 4 | n = 15 | n = 5 | n = 6 |
| Target Gene | Forward Primer | Reverse Primer |
|---|---|---|
| 18S | CTCAACACGGGAAACCTCAC | CGCTCCACCAACTAAGAACG |
| α-SMA | TTCCTTCGTGACTACTGCCG | GCTGTTATAGGTGGTTTCGTGG |
| C3 | ATCCAGACAGACCAGACCATCT | AGGATGACGACTGTCTTGCC |
| Col1a1 | GACGCATGGCCAAGAAGACA | CATTGCACGTCATCGCACAC |
| FN1 | CAGACCTACCCAGGCACAAC | CAGCGACCCGTAGAGGTTTT |
| HIF-1α | GGAGCCTTAACCTGTCTGCC | TGCTCCGTTCCATTCTGTTCA |
| HIF-2α | CCCTGCTGTCCTGCCTTATC | CATAGGCAGAGCGTCCAAGT |
| IL-6 | CAAAGCCAGAGTCCTTCAGAGA | GGTCTTGGTCCTTAGCCACTC |
| MCP-1 | TCACTGAAGCCAGCTCTCTCT | TCTTGTAGCTCTCCAGCCTACT |
| LCN-2 | ACGGACTACAACCAGTTCGC | AATGCATTGGTCGGTGGGG |
| NRF2 | CCTCACCTCTGCTGCAAGTA | GCTCATAGTCCTTCTGTCGCT |
| TNF-α | AAATGGCCTCCCTCTCATCA | AGATAGCAAATCGGCTGACG |
| TGF-β | CAACAATTCCTGGCGTTACCTTGG | GAAAGCCCTGTATTCCGTCTCCTT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tod, P.; Bukosza, E.N.; Róka, B.; Kaucsár, T.; Fintha, A.; Krenács, T.; Szénási, G.; Hamar, P. Post-Ischemic Renal Fibrosis Progression Is Halted by Delayed Contralateral Nephrectomy: The Involvement of Macrophage Activation. Int. J. Mol. Sci. 2020, 21, 3825. https://doi.org/10.3390/ijms21113825
Tod P, Bukosza EN, Róka B, Kaucsár T, Fintha A, Krenács T, Szénási G, Hamar P. Post-Ischemic Renal Fibrosis Progression Is Halted by Delayed Contralateral Nephrectomy: The Involvement of Macrophage Activation. International Journal of Molecular Sciences. 2020; 21(11):3825. https://doi.org/10.3390/ijms21113825
Chicago/Turabian StyleTod, Pál, Eva Nora Bukosza, Beáta Róka, Tamás Kaucsár, Attila Fintha, Tibor Krenács, Gábor Szénási, and Péter Hamar. 2020. "Post-Ischemic Renal Fibrosis Progression Is Halted by Delayed Contralateral Nephrectomy: The Involvement of Macrophage Activation" International Journal of Molecular Sciences 21, no. 11: 3825. https://doi.org/10.3390/ijms21113825
APA StyleTod, P., Bukosza, E. N., Róka, B., Kaucsár, T., Fintha, A., Krenács, T., Szénási, G., & Hamar, P. (2020). Post-Ischemic Renal Fibrosis Progression Is Halted by Delayed Contralateral Nephrectomy: The Involvement of Macrophage Activation. International Journal of Molecular Sciences, 21(11), 3825. https://doi.org/10.3390/ijms21113825