Positional Correlation Natural Vector: A Novel Method for Genome Comparison


Abstract
:1. Introduction
2. Results
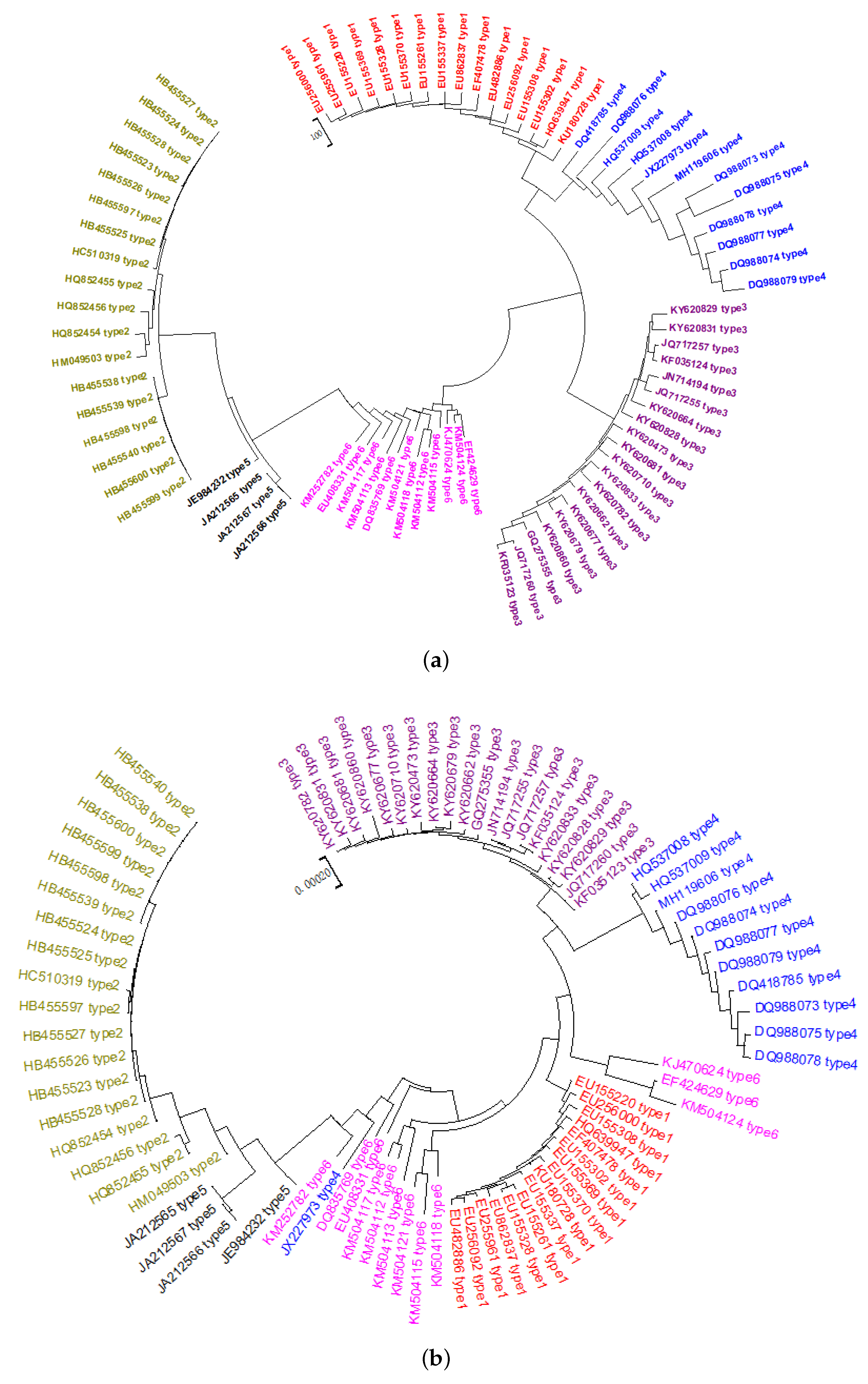
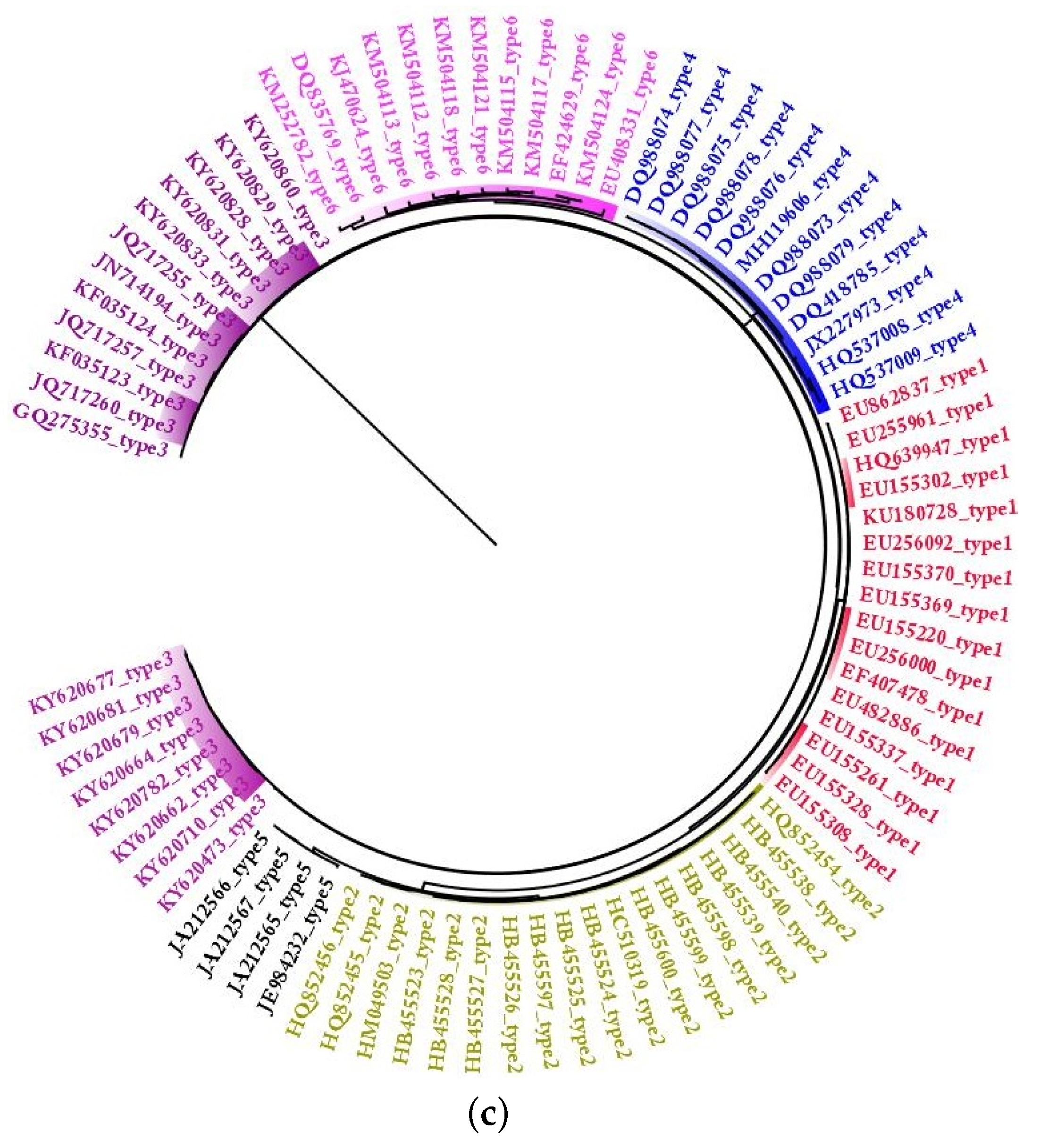
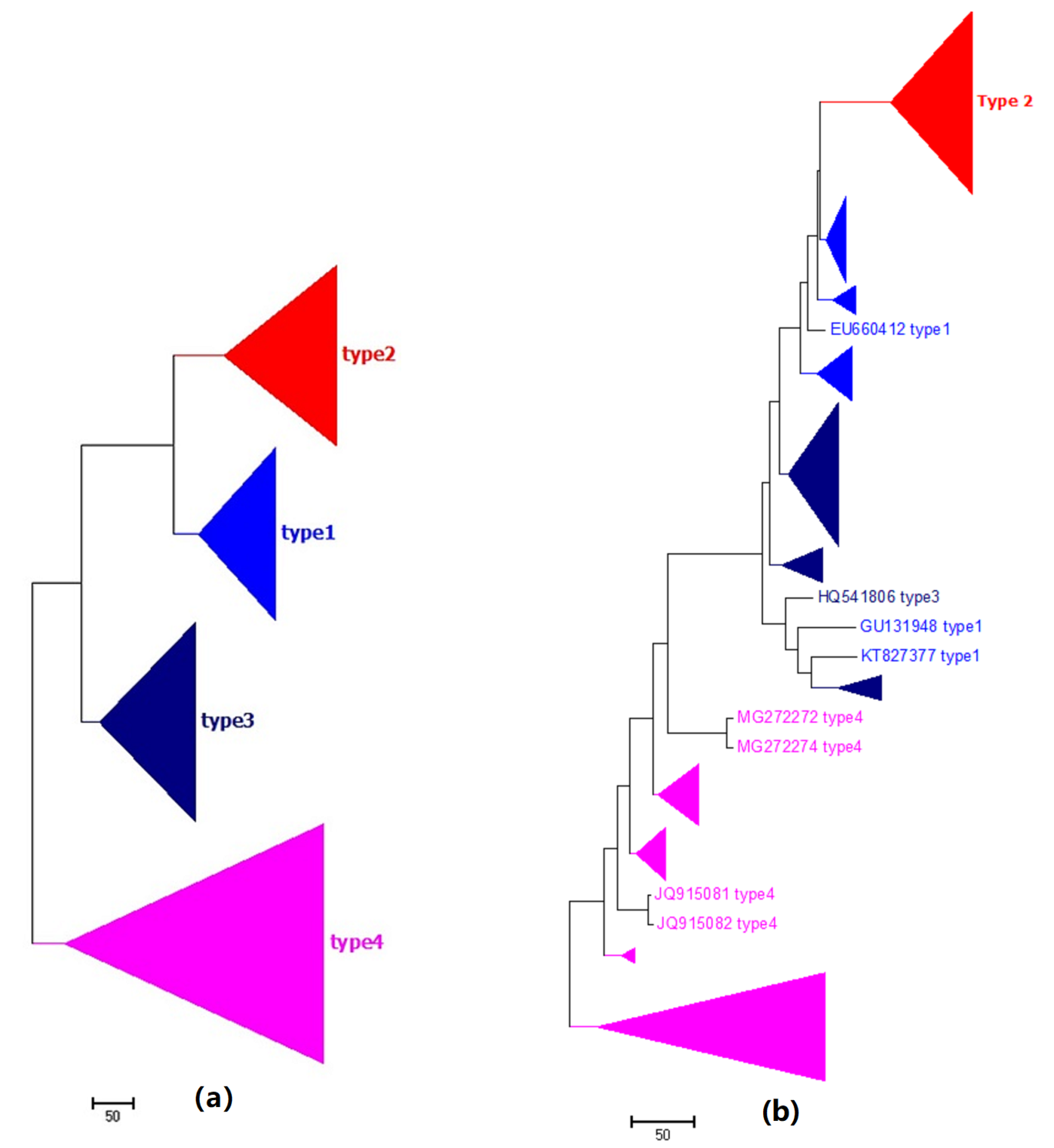
2.1. Phylogeny of HCV
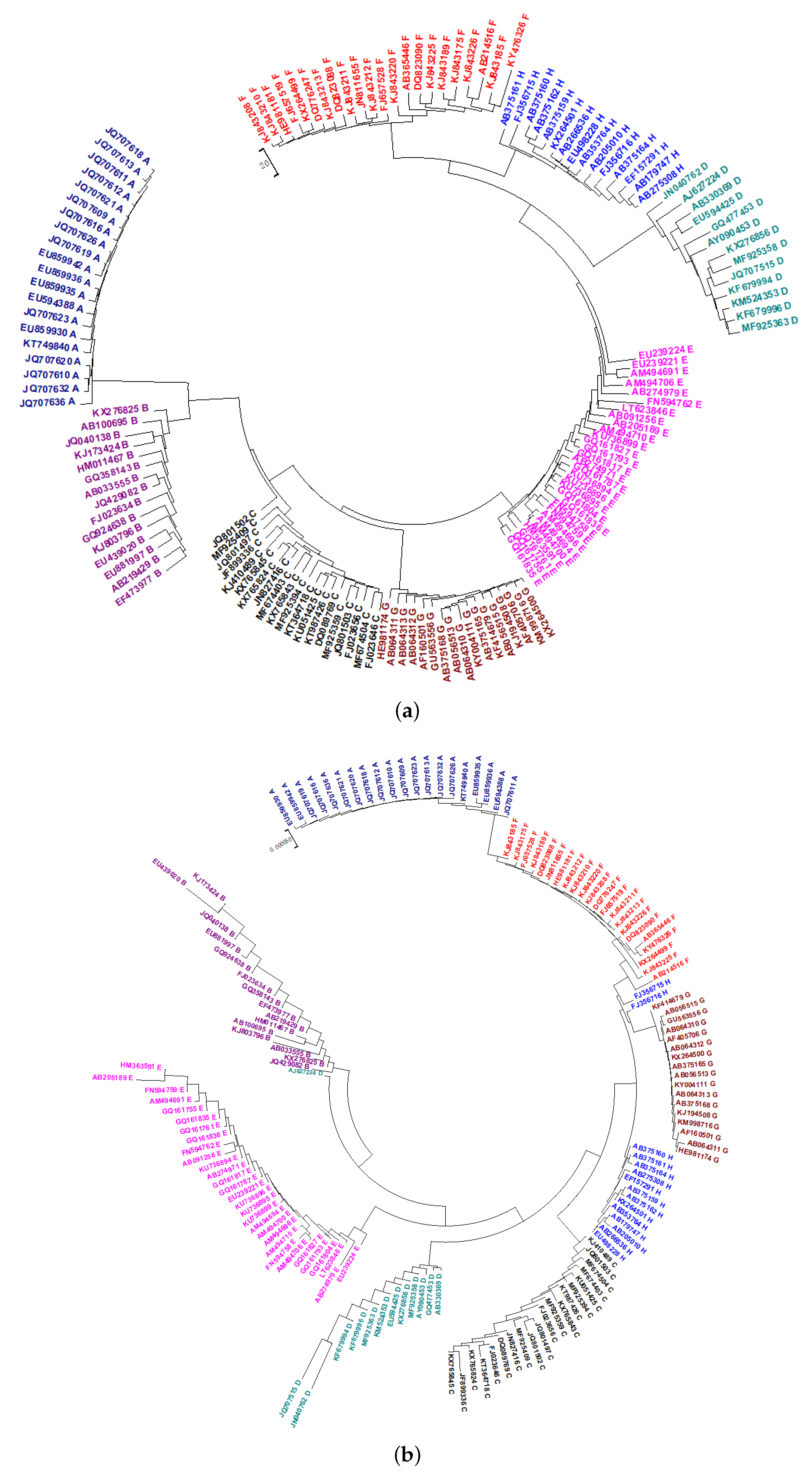
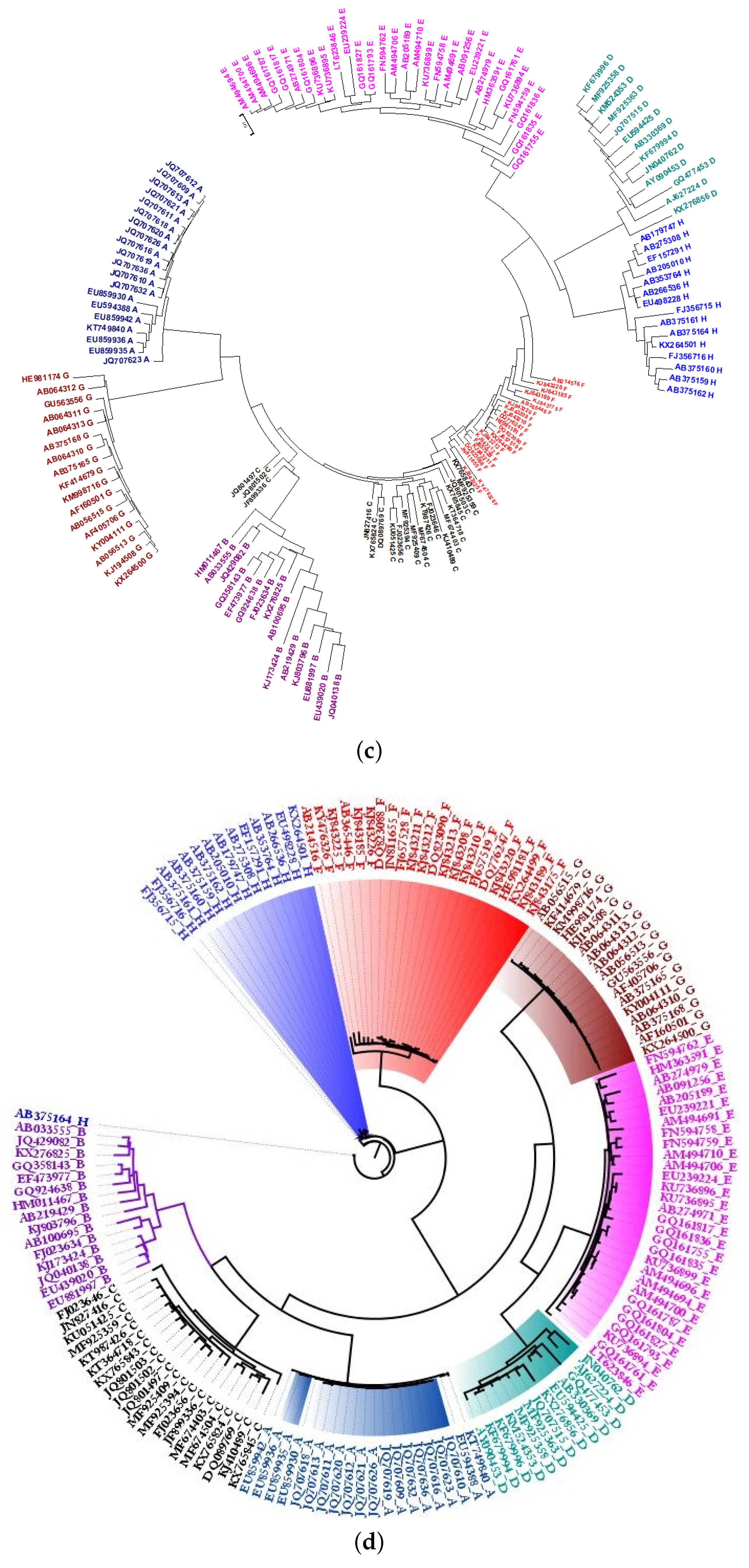
2.2. Phylogeny of HBV
2.3. Phylogeny of DENV
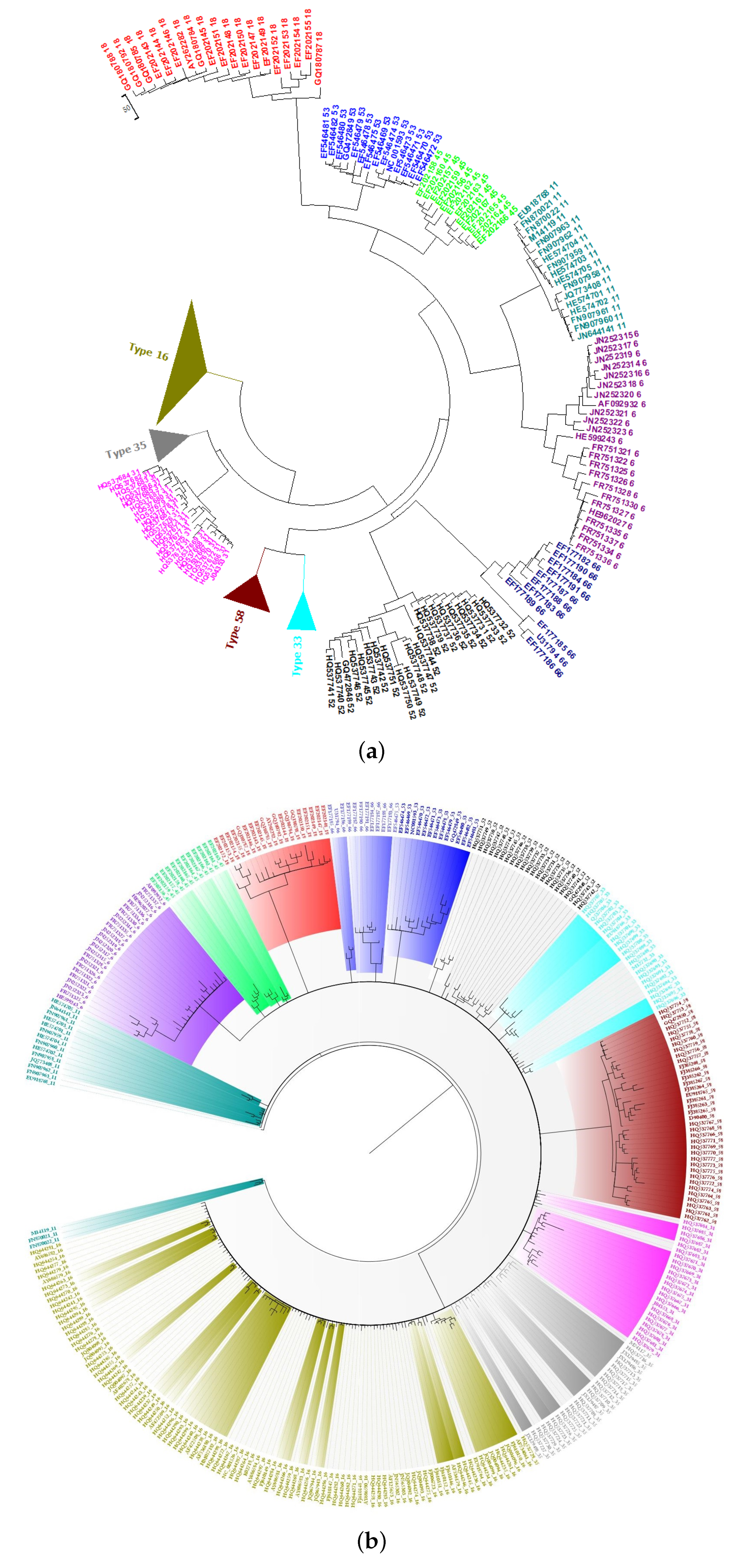
2.4. Phylogeny of HPV
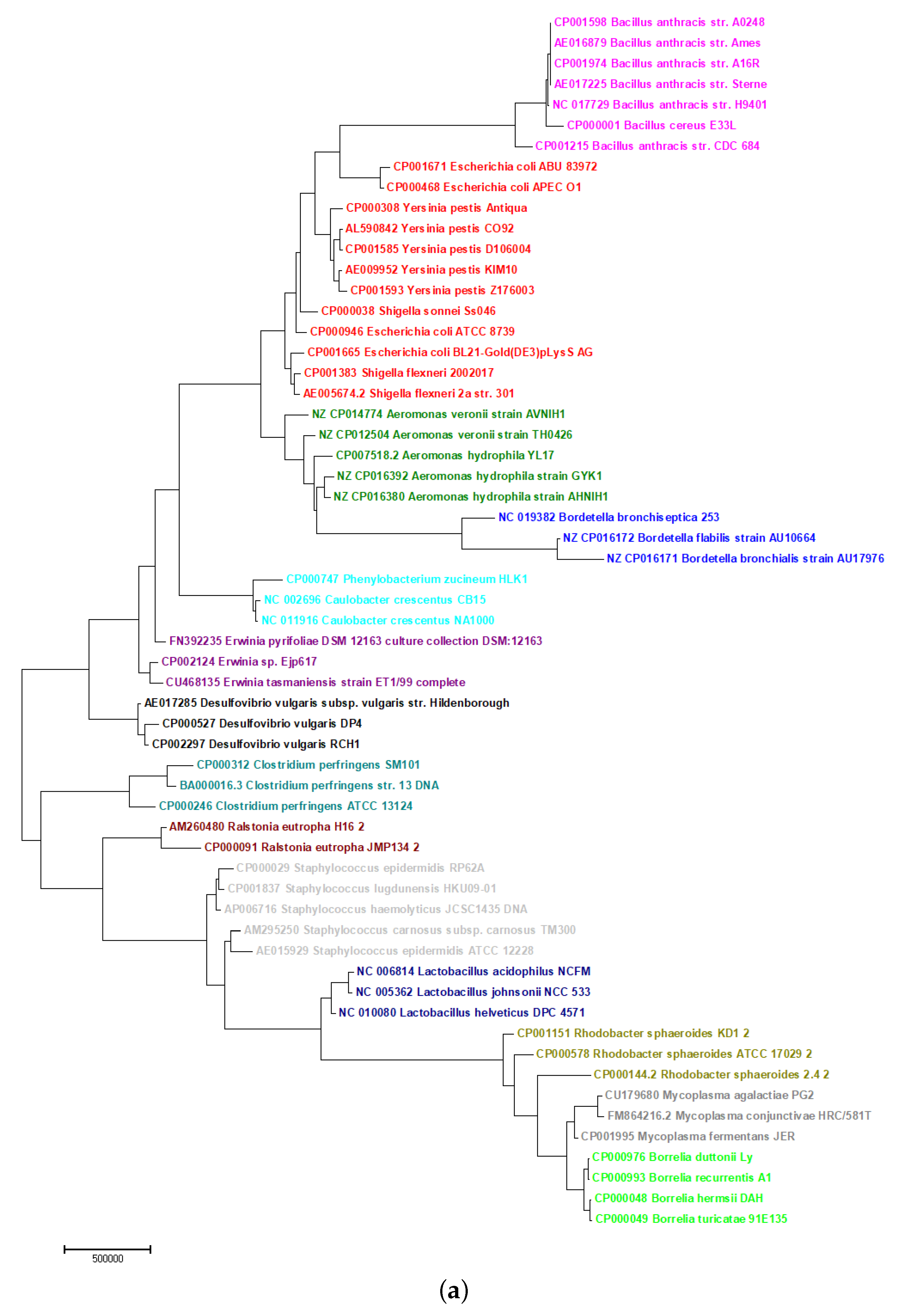
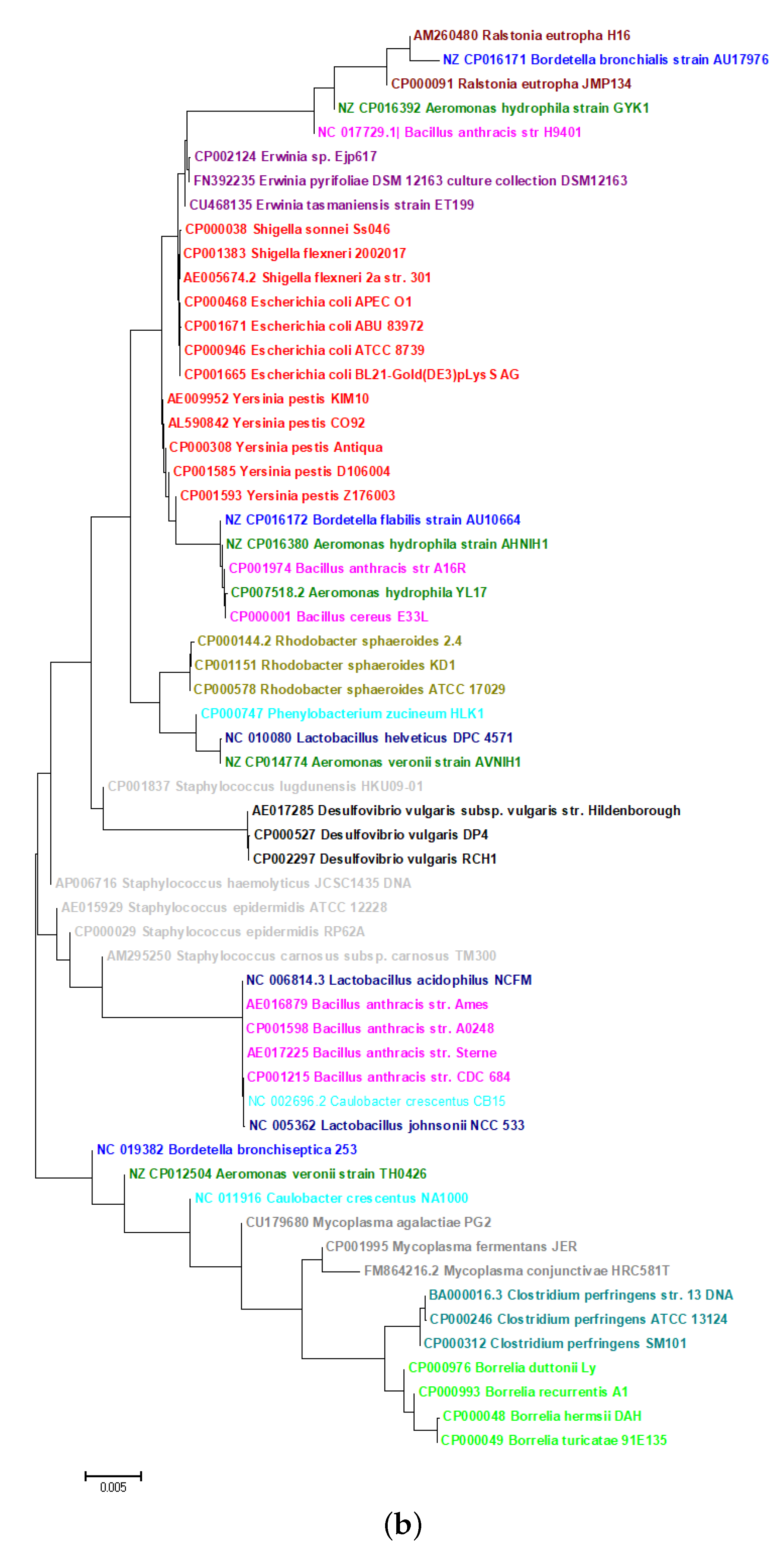
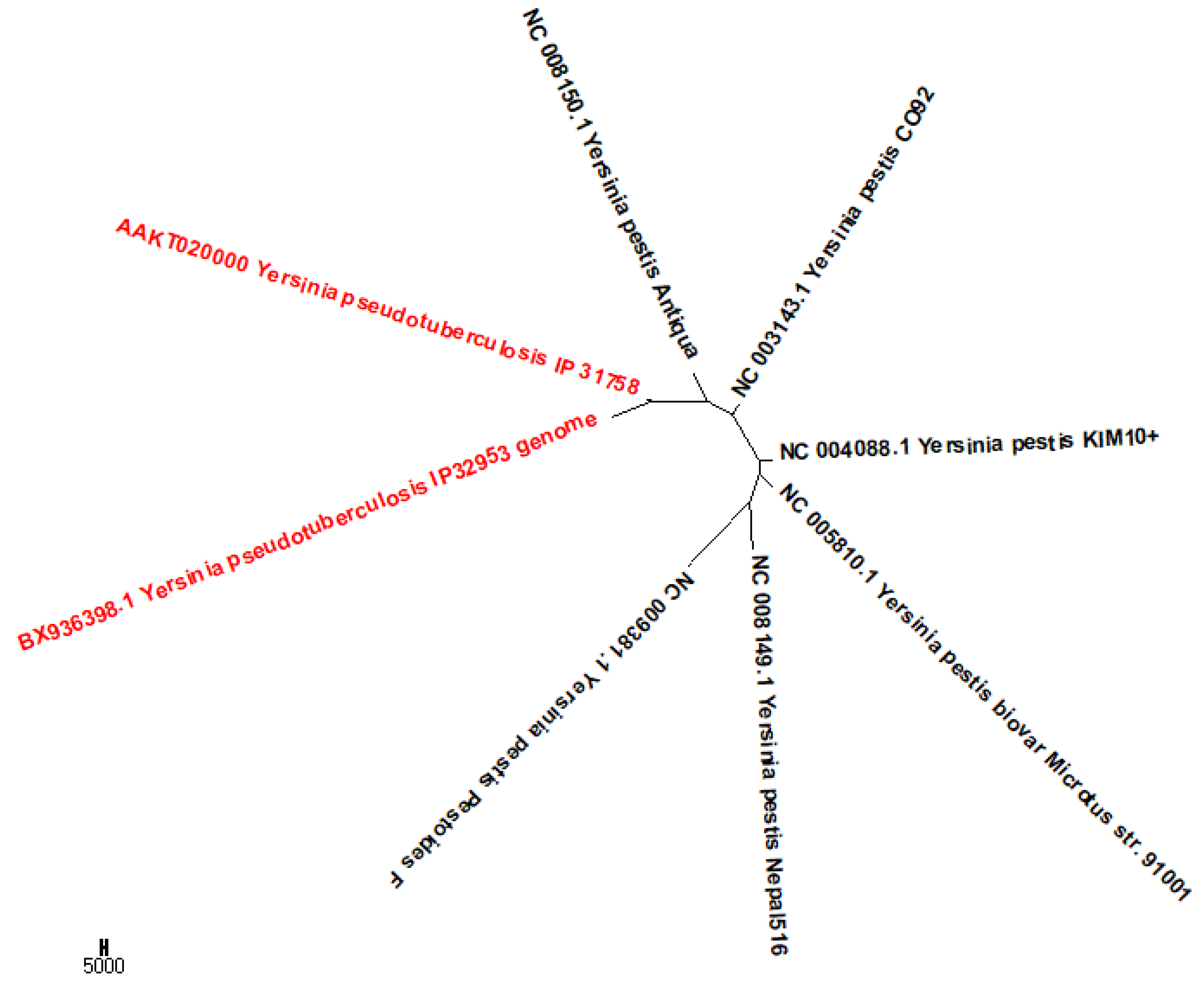
2.5. Phylogeny of Bacteria
2.6. Classification
3. Discussion
4. Materials and Methods
4.1. Dataset
4.1.1. HCV
4.1.2. HBV
4.1.3. DENV
4.1.4. HPV
4.1.5. Bacteria
4.2. Positional Distribution
- (1)
- When , namely, , so .
- (2)
- When , namely, , thus .
- (3)
- When , namely, , .
4.3. Position Correlation Vector
4.3.1. Average Positional Distribution
4.3.2. Positional Covariance
4.3.3. Positional variance
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFKS | Alignment-Free-Kmer-Statistics |
| DENV | Dengue virus |
| HCV | Hepatitis C virus |
| HBV | Hepatitis B virus |
| HMM | hidden Markov model |
| HPV | Human papillomavirus |
| PCNV | Positional Correlation Natural Vector |
| NV | Natural Vector |
| ViPR | Virus Pathogen Database and Analysis Resource |
| FFP | Feature Frequency Profiles |
References
- Fredrik, R.; Maxim, T.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Liu, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar]
- Bernard, G.; Chan, C.X.; Chan, Y.-B.; Chua, X.-Y.; Cong, Y.; Hogan, J.M. Alignment-free inference of hierarchical and reticulate phylogenomic relationships. Brief. Bioinform. 2017, 2, 2. [Google Scholar]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. AAlignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 1, 186. [Google Scholar] [CrossRef] [Green Version]
- Zielezinski, A.; Girgis, H.Z.; Bernard, G. Benchmarking of alignment-free sequence comparison methods. Genome Biol. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Vinga, S. Information theory applications for biological sequence analysis. Genome Biol. 2014, 15, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Almeida, J.S. Sequence analysis by iterated maps, a review. Brief. Bioinform. 2014, 15, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Yau, S.S.-T. An improved model for whole genome phylogenetic analysis by Fourier transform. J. Theor. Biol. 2015, 382, 99–110. [Google Scholar] [CrossRef]
- Jeffrey, H.J. Chaos game representation of gene structure. NAR 1990, 18, 2163–2170. [Google Scholar]
- Deng, M.; Yu, C.L.; Liang, Q.; He, R.L.; Yau, S.S.-T. A novel method of characterizing genetic sequences: Genome space with biological distance and applications. PLoS ONE 2011, 31, e17293. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, L.; Lucy, H.R.; Yau, S.S.-T. A novel fast vector method for genetic sequence comparison. Sci. Rep. 2017, 7, 12226. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Bai, X.; Lu, Y.Y.; Tang, K.; Wang, Y.; Reinert, G. Alignment-free sequence analysis and applications. Annu. Rev. Biomed. Data Sci. 2018, 1, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.H.A.J.; Juna, S.-R.; Simsa, G.E.S.; Kim, S.-H. Whole-proteome phylogeny of large dsDNA virus families by an alignment-free method. PNAS 2009, 106, 12826–12831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Q.; Yang, Y.C.; Wang, T.M. Markov model plus k-word distributions: A synergy that produces novel statistical measures for sequence comparison. Bioinformatics 2012, 20, 2296–2302. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Hao, B.L. CVTree update: A newly designed phylogenetic study platform using composition vectors and whole genomes. NAR 2009, 37 (Suppl. 2), W174–W178. [Google Scholar] [CrossRef] [Green Version]
- Hatje, K.; Kollmar, M. A phylogenetic analysis of the brassicales clade based on an alignment-free sequence comparison method. Front. Plant Sci. 2012, 3, 192. [Google Scholar] [CrossRef] [Green Version]
- Luczak, B.B.; James, B.T.; Girgis, H.Z. A survey and evaluations of histogram-based statistics in alignment-ree sequence comparison. Brief. Bioinform. 2017, 20, 1222–1237. [Google Scholar] [CrossRef] [Green Version]
- Leimeister, C.-A.; Boden, M.; Horwege, S.; Lindner, S.; Morgenstern, B. Fast alignment-free sequence comparison using spaced-word frequencies. Bioinformatics 2014, 30, 1991–1999. [Google Scholar] [CrossRef] [Green Version]
- Leimeister, C.-A.; Sohrabi-Jahromi, S.; Morgenstern, B. Fast and accurate phylogeny reconstruction using filtered spaced-word matches. Bioinformatics 2017, 33, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.D.; Webers, C.; Ong, C.S.; Borevitz, J.; Warthmann, N. kWIP: The k-mer weighted inner product, a de novo estimator of genetic similarity. PLoS Comput. Biol. 2017, 13, e1005727. [Google Scholar] [CrossRef] [Green Version]
- Blaisdell, B.E. Average values of a dissimilarity measure not requiring sequence alignment are twice the averages of conventional mismatch counts requiring sequence alignment for a computer-generated model system. J. Mol. Evol. 1989, 29, 538–547. [Google Scholar] [CrossRef]
- Sims, G.E.; Jun, S.R.; Wu, G.A.; Kim, S.H. Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. PNAS 2009, 106, 2677–2682. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.-J. Hidden Markov Models and their Applications in Biological Sequence Analysis. Curr. Genom. 2009, 10, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.Y.; Gu, Z.P.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus pathogen database and analy-sis resource (vipr): A comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef] [Green Version]
- Kramer, O. K-Nearest Neighbors. Dimensionality Reduction with Unsupervised Nearest Neighbors; Springer: Berlin/Heidelberg, Germany, 2013; pp. 13–23. [Google Scholar]
- Koski, L.B.; Golding, G.B. The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 2001, 52, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Echeverría, N.; Moratorio, G.; Cristina, J.; Moreno, P. Hepatitis C virus genetic variability and evolution. World J. Hepatol. 2015, 7, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Campo, D.S.; Dimitrova, Z.; Mitchell, R.J.; Lara, J.; Khudyakov, Y. Coordinated evolution of the hepatitis C virus. PNAS 2008, 105, 9685–9690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, M.-F.; Chen, D.-S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.-L. Hepatitis B virus infection. Nat. Rev. Dis.Prim. 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.Z.; Klunk, J.; Fornaciari, G.; Giuffra, V.; Duchěne, S.; Duggan, A.T.; Poinar, D.; Douglas, M.W.; Eden, J.-S.; Holmes, E.C.; et al. Correction: The paradox of hbv evolution as revealed from a 16th century mummy. PLoS Pathog. 2012, 14, e1006887. [Google Scholar] [CrossRef]
- Hayer, J.; Jadeau, F.; Deléage, G.; Kay, A.; Zoulim, F.; Combet, C. HBVdb: A knowledge database for Hepatitis B Virus. NAR 2013, 41, D566–D570. [Google Scholar] [CrossRef] [Green Version]
- Norder, H.; Hammas, B.; Lee, S.D.; Bile, K.; Courouc, A.M.; Mushahwar, I.K.; Magnius, L.O. Genetic relatedness of hepatitis B viral strains of diverse geographical origin and natural variations in the primary structure of the surface antigen. J. Gen. Virol. 1993, 74, 1341–1348. [Google Scholar] [CrossRef]
- Sirisena, P.D.N.N.; Noordeen, F. Evolution of dengue in Sri Lanka–changes in the virus, vector, and climate. Int. J. Infect. Dis. 2014, 19, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, T.K.; Ghebremariam, S.L.; Gresh, L.; Gordon, A.; Halloran, M.E.; Katzelnick, L.C.; Rojas, D.P.; Kuan, G.; Balmaseda, A.; Sugimoto, J.; et al. Effects of infection history on dengue virus infection and pathogenicity. Nat. Commun. 2019, 10, 1246. [Google Scholar] [CrossRef]
- Eom, J.H.; Park, S.B.; Zhang, B.T. Genetic Mining of DNA Sequence Structures for Effective Classification of the Risk Types of Human Papillomavirus (HPV). Lect. Notes Comput. Sci. 2004, 3316, 1334–1343. [Google Scholar]
- Smith, J.S.; Lindsay, L.; Hoots, B.; Keys, J.; Franceschi, S.; Winer, R.; Clifford, G.M. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: A meta-analysis update. Int. J. Cancer 2007, 121, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | HCV | HBV | Dengue | HPV | Bacteria |
|---|---|---|---|---|---|
| (82) | (152) | (330) | (326) | (59) | |
| PCNV | 0.33s | 0.27s | 0.66s | 0.78s | 53.71s |
| Bayesian | 1097s | 263s | 217,353s | 217,512s | ∼ |
| inference | |||||
| FFP | 11.11s | 0.38s | 49.40s | 35.00s | larger than |
| (k = 6) | (k = 5) | (k = 6) | (k = 6) | 1 day (k = 11) | |
| AFKS | 70.21s | 29.62s | 429.87s | 413.79s | larger than |
| (k = 5) | (k = 4) | (k = 5) | (k = 5) | 4 day (k = 9) | |
| Muscle | 753s | 155s | 3740s | 4002s | ∼ |
| Nu- | Sens | Sens | Sens | Spec | Spec | Spec | Acc | Acc | Acc | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | mber | PCNV | FFP | AFKS | PCNV | FFP | AFKS | PCNV | FFP | AFKS | |
| (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | (%) | |||
| HCV (82) | type1 | 16 | 100 | 62.5 | 50.0 | 100 | 87.9 | 86.4 | 100 | 62.5 | 50.0 |
| type2 | 18 | 100 | 55.6 | 94.4 | 100 | 93.8 | 98.4 | 100 | 55.6 | 94.4 | |
| type3 | 20 | 100 | 80.0 | 90.0 | 100 | 93.5 | 96.8 | 100 | 80.0 | 90.0 | |
| type4 | 12 | 100 | 50.0 | 33.3 | 100 | 97.1 | 90.0 | 100 | 50.0 | 33.3 | |
| type5 | 4 | 100 | 50.0 | 75.0 | 100 | 96.2 | 97.4 | 100 | 50.0 | 75.0 | |
| type6 | 12 | 100 | 50.0 | 83.3 | 100 | 91.4 | 98.6 | 100 | 50.0 | 83.3 | |
| Ave. | 100 | 58.0 | 71.0 | 100 | 93.3 | 94.6 | 100 | 58.0 | 71.0 | ||
| HBV (152) | A | 20 | 100 | 100 | 100 | 100 | 100 | 99.2 | 100 | 100 | 100 |
| B | 15 | 100 | 100 | 40.0 | 100 | 100 | 96.4 | 100 | 100 | 40.0 | |
| C | 20 | 100 | 100 | 70.0 | 100 | 100 | 96.2 | 100 | 100 | 70.0 | |
| D | 13 | 100 | 100 | 76.9 | 100 | 100 | 97.1 | 100 | 100 | 76.9 | |
| E | 30 | 100 | 100 | 90.0 | 100 | 100 | 97.5 | 100 | 100 | 90.0 | |
| F | 22 | 100 | 100 | 72.7 | 100 | 100 | 93.8 | 100 | 100 | 72.7 | |
| G | 17 | 100 | 100 | 94.1 | 100 | 100 | 99.3 | 100 | 100 | 94.1 | |
| H | 15 | 100 | 100 | 80.0 | 100 | 100 | 97.1 | 100 | 100 | 80.0 | |
| Ave. | 100 | 100 | 78.0 | 100 | 100 | 97.1 | 100 | 100 | 78.0 | ||
| Dengue (330) | type1 | 72 | 100 | 100 | 76.4 | 100 | 100 | 93.4 | 100 | 100 | 76.4 |
| type2 | 75 | 100 | 100 | 73.3 | 100 | 100 | 93.3 | 100 | 100 | 73.3 | |
| type3 | 83 | 100 | 100 | 78.3 | 100 | 100 | 92.7 | 100 | 100 | 78.3 | |
| type4 | 100 | 100 | 100 | 87.0 | 100 | 100 | 93.0 | 100 | 100 | 87.0 | |
| Ave. | 100 | 100 | 78.8 | 100 | 100 | 93.1 | 100 | 100 | 78.8 | ||
| HPV (326) | 6 | 24 | 100 | 100 | 75.0 | 100 | 100 | 97.7 | 100 | 100 | 75.0 |
| 11 | 17 | 100 | 100 | 100 | 100 | 100 | 99.7 | 100 | 100 | 100 | |
| 16 | 99 | 100 | 100 | 92.9 | 100 | 100 | 96.5 | 100 | 100 | 92.9 | |
| 18 | 19 | 100 | 100 | 94.7 | 100 | 100 | 100 | 100 | 100 | 94.7 | |
| 31 | 23 | 100 | 100 | 82.6 | 100 | 100 | 99.0 | 100 | 100 | 82.6 | |
| 33 | 22 | 100 | 100 | 86.4 | 100 | 100 | 99.7 | 100 | 100 | 86.4 | |
| 35 | 26 | 100 | 100 | 88.5 | 100 | 100 | 99.0 | 100 | 100 | 88.5 | |
| 45 | 12 | 100 | 100 | 83.3 | 100 | 100 | 99.7 | 100 | 100 | 83.3 | |
| 52 | 22 | 100 | 100 | 81.8 | 100 | 100 | 98.4 | 100 | 100 | 81.8 | |
| 53 | 14 | 100 | 100 | 85.7 | 100 | 100 | 98.7 | 100 | 100 | 85.7 | |
| 58 | 37 | 100 | 100 | 94.6 | 100 | 100 | 99.7 | 100 | 100 | 94.6 | |
| 66 | 11 | 100 | 100 | 90.9 | 100 | 100 | 99.7 | 100 | 100 | 90.9 | |
| Ave. | 100 | 100 | 88.0 | 100 | 100 | 99.0 | 100 | 100 | 88.0 |
| Dataset | Number | Min | Median | Mean | Max |
|---|---|---|---|---|---|
| (bp) | (bp) | (bp) | (bp) | ||
| HCV | 82 | 8957 | 9442 | 9427 | 9666 |
| HBV | 152 | 10161 | 10669 | 10606 | 10780 |
| Dengue | 330 | 10,161 | 10,669 | 10,606 | 10,780 |
| HPV | 326 | 7814 | 7,905 | 7895 | 8051 |
| Bacteria | 59 | 846,214 | 4,016,947 | 3,610,938 | 5,966,919 |
| Sequence | A | C | T | G | G | C | A | A | T |
|---|---|---|---|---|---|---|---|---|---|
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| 0 | |||||||||
| 0 | 0 | ||||||||
| 0 | 0 | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Dong, R.; He, R.L.; Yau, S.S.-T. Positional Correlation Natural Vector: A Novel Method for Genome Comparison. Int. J. Mol. Sci. 2020, 21, 3859. https://doi.org/10.3390/ijms21113859
He L, Dong R, He RL, Yau SS-T. Positional Correlation Natural Vector: A Novel Method for Genome Comparison. International Journal of Molecular Sciences. 2020; 21(11):3859. https://doi.org/10.3390/ijms21113859
Chicago/Turabian StyleHe, Lily, Rui Dong, Rong Lucy He, and Stephen S.-T. Yau. 2020. "Positional Correlation Natural Vector: A Novel Method for Genome Comparison" International Journal of Molecular Sciences 21, no. 11: 3859. https://doi.org/10.3390/ijms21113859
APA StyleHe, L., Dong, R., He, R. L., & Yau, S. S. -T. (2020). Positional Correlation Natural Vector: A Novel Method for Genome Comparison. International Journal of Molecular Sciences, 21(11), 3859. https://doi.org/10.3390/ijms21113859