Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease





{kind=link}
{kind=link}
Abstract
:1. Introduction
2. SIRT1 in Liver Metabolism
3. SIRT1 in a Liver Metabolic Disorder: NAFLD (Non-Alcoholic Fatty Liver Disease)
4. SIRT1 in NF-κB Mediated Inflammation
5. Hepatic Inflammation
5.1. NF-κB in Hepatocytes: Survival and Proliferation
5.2. NF-κB in Kupffer Cells (KCs): Inflammation
5.3. NF-κB in Hepatic Stellate Cells (HSCs): Activation, Survival and Inflammation
6. Upstream Regulators of SIRT1
6.1. IRF9/PPAR-α
6.2. NAMPT/NMNAT
6.3. HuR
6.4. AMPK
6.5. PARP1
6.6. CK2
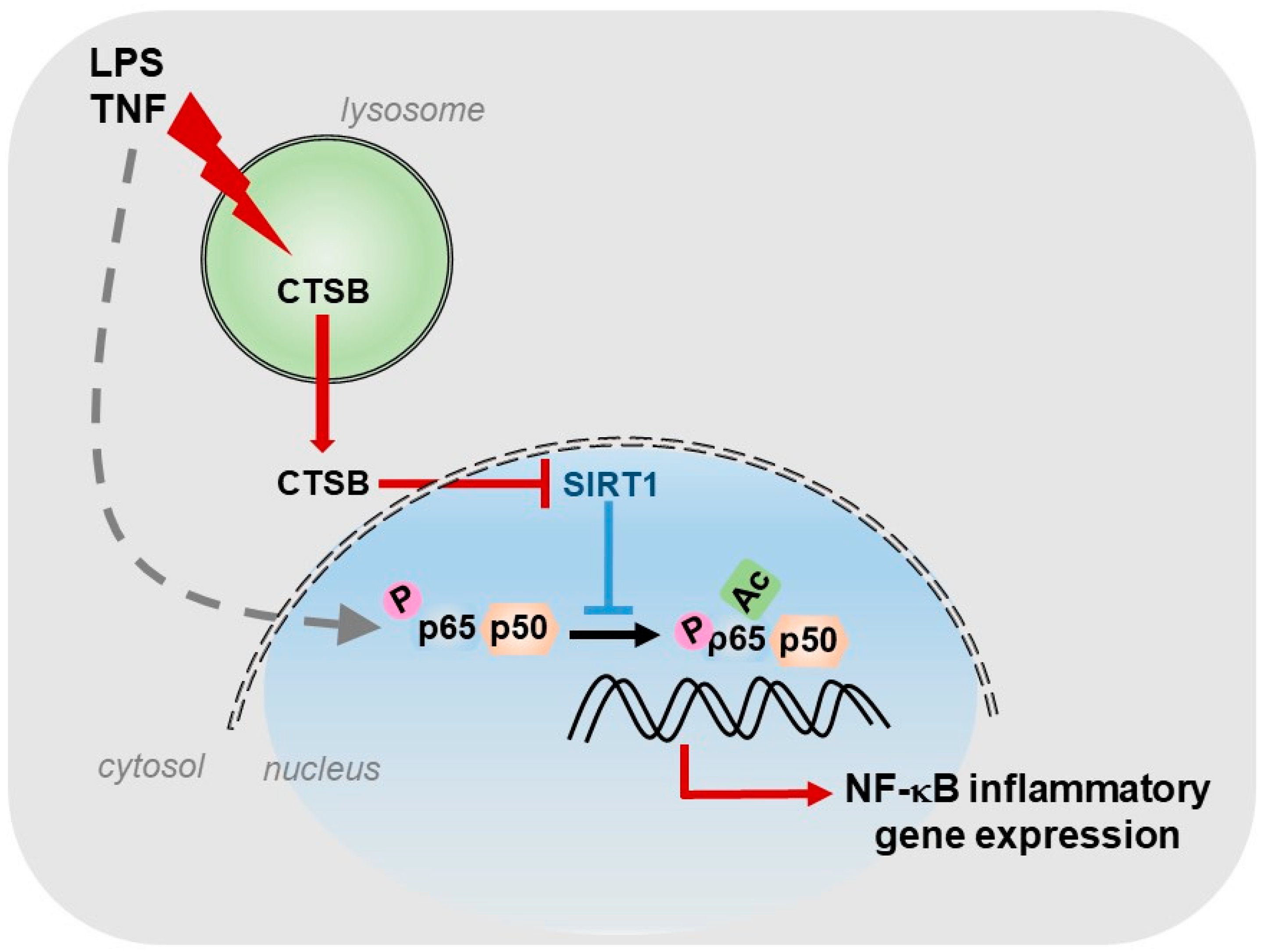
6.7. Cathepsins
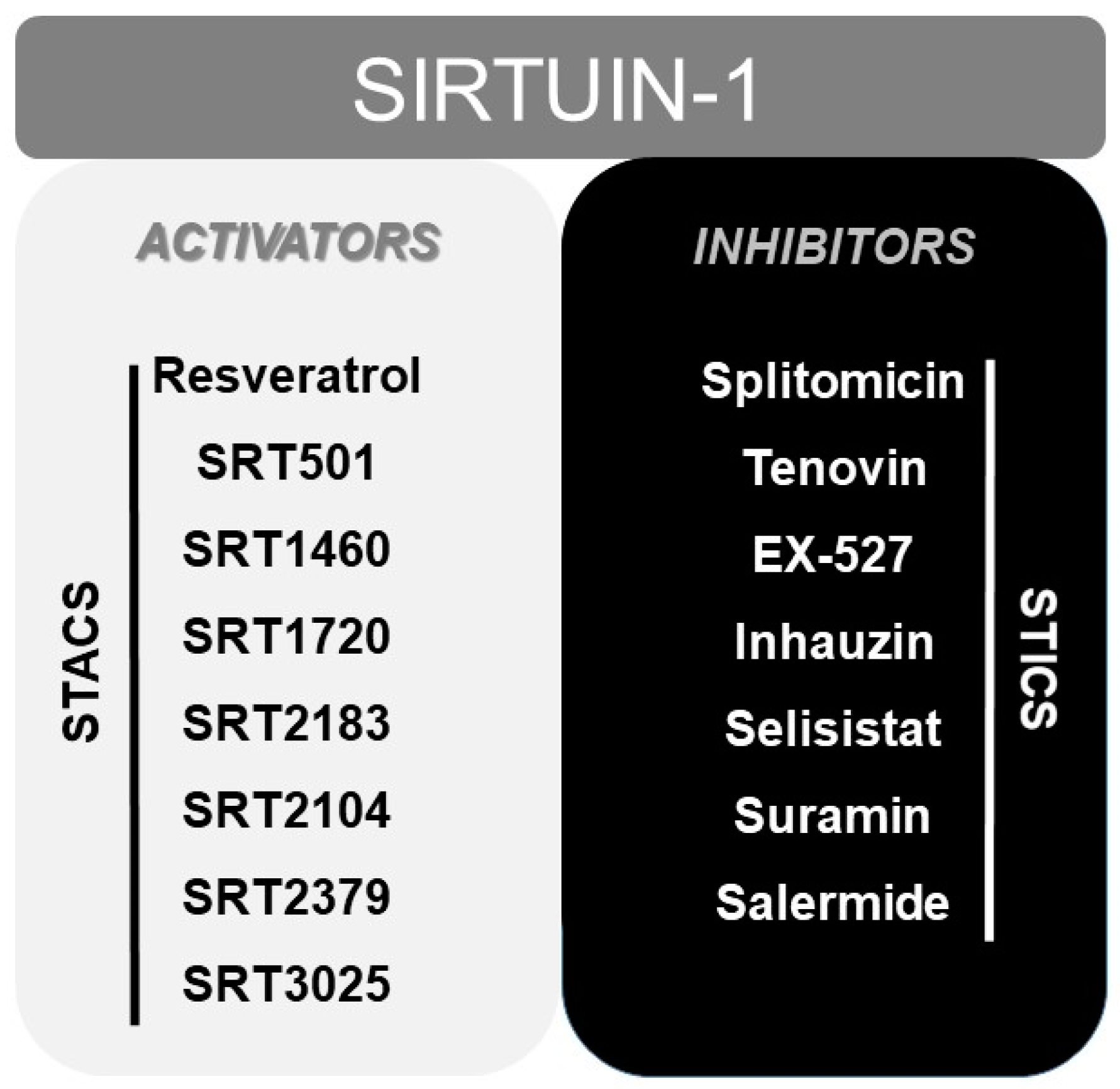
7. Other Modulators of SIRT1: From Resveratrol to STACs (Sirtuin Activating Compounds)
8. Other Considerations: Anti-Oxidant Properties of SIRT1
9. Conclusions
Funding
Conflicts of Interest
References
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [PubMed]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [Green Version]
- Ong, A.L.C.; Ramasamy, T.S. Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res. Rev. 2018, 43, 64–80. [Google Scholar]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef]
- Higashida, K.; Kim, S.H.; Jung, S.R.; Asaka, M.; Holloszy, J.O.; Han, D.-H. Effects of Resveratrol and SIRT1 on PGC-1α Activity and Mitochondrial Biogenesis: A Reevaluation. PLoS Biol. 2013, 11, e1001603. [Google Scholar] [CrossRef] [Green Version]
- De Mingo, Á.; De Gregorio, E.; Moles, A.; Tarrats, N.; Tutusaus, A.; Colell, A.; Fernandez-Checa, J.C.; Morales, A.; Marí, M. Cysteine cathepsins control hepatic NF-κB-dependent inflammation via sirtuin-1 regulation. Cell Death Dis. 2016, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rajamohan, S.B.; Pillai, V.B.; Gupta, M.; Sundaresan, N.R.; Birukov, K.G.; Samant, S.; Hottiger, M.O.; Gupta, M.P. SIRT1 Promotes Cell Survival under Stress by Deacetylation-Dependent Deactivation of Poly(ADP-Ribose) Polymerase 1. Mol. Cell Biol. 2009, 29, 4116–4129. [Google Scholar]
- Mendes, K.L.; Lelis, D.D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [PubMed]
- Guarente, L. Calorie Restriction and Sirtuins Revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [PubMed] [Green Version]
- Chalkiadaki, A.; Guarente, L. Metabolic signals regulate SIRT1 expression. EMBO Rep. 2011, 12, 985–986. [Google Scholar]
- Yamagata, K.; Yoshizawa, T. Chapter Four—Transcriptional Regulation of Metabolism by SIRT1 and SIRT7. In Transcriptional Gene Regulation in Health and Disease; Loos, F., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 143–166. [Google Scholar]
- Liu, Y.; Dentin, R.; Chen, D.; Hedrick, S.; Ravnskjaer, K.; Schenk, S.; Milne, J.; Meyers, D.J.; Cole, P.; Yates, J., 3rd; et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 2008, 456, 269–273. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Frescas, D.; Valenti, L.; Accili, D. Nuclear trapping of the forkhead transcription factor FoxO1 via sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005, 280, 20589–20595. [Google Scholar] [CrossRef] [Green Version]
- Noriega, L.G.; Feige, J.N.; Canto, C.; Yamamoto, H.; Yu, J.; Herman, M.A.; Mataki, C.; Kahn, B.B.; Auwerx, J. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011, 12, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Yang, F.; Jiang, K.; Ji, J.Y.; Watts, J.L.; Purushotham, A.; Boss, O.; Hirsch, M.L.; Ribich, S.; Smith, J.J.; et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010, 24, 1403–1417. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 Deacetylates and Positively Regulates the Nuclear Receptor LXR. Mol. Cell. 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Kemper, J.K.; Xiao, Z.; Ponugoti, B.; Miao, J.; Fang, S.; Kanamaluru, D.; Tsang, S.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D. FXR Acetylation Is Normally Dynamically Regulated by p300 and SIRT1 but Constitutively Elevated in Metabolic Disease States. Cell Metab. 2009, 10, 392–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 46, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Singal, A.K. Emerging medical therapies for non-alcoholic fatty liver disease and for alcoholic hepatitis. Transl. Gastroenterol. Hepatol. 2019, 4, 53. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)—Pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar]
- Rodgers, J.T.; Puigserver, P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866. [Google Scholar]
- Wang, R.-H.; Li, C.; Deng, C.-X. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 2010, 6, 682–690. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Liu, Y.; Fu, Y.; Liu, X.; Zhou, X. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- Mariani, S.; Fiore, D.; Basciani, S.; Persichetti, A.; Contini, S.; Lubrano, C.; Salvatori, L.; Lenzi, A.; Gnessi, L. Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 2015, 49, 711–716. [Google Scholar] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [PubMed]
- Zhang, Z.; Lowry, S.F.; Guarente, L.; Haimovich, B. Roles of SIRT1 in the acute and restorative phases following induction of inflammation. J. Biol. Chem. 2010, 285, 41391–41401. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Karin, M. NF-kappaB and STAT3—Key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.P.; Carmody, R.J. Chapter Two—NF-κB and the Transcriptional Control of Inflammation. In Transcriptional Gene Regulation in Health and Disease; Loos, F., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 41–84. [Google Scholar]
- Schmitz, M.L.; Mattioli, I.; Buss, H.; Kracht, M. NF-kappaB: A multifaceted transcription factor regulated at several levels. Chembiochem 2004, 5, 1348–1358. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-kappaB in the liver—Linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-κB Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013, 25, 1939–1948. [Google Scholar] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-D.; Tajkhorshid, E.; Chen, L.-F. Functional Interplay between Acetylation and Methylation of the RelA Subunit of NF-κB. Mol. Cell Biol. 2010, 30, 2170–2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothgiesser, K.M.; Fey, M.; Hottiger, M.O. Acetylation of p65 at lysine 314 is important for late NF-kappaB-dependent gene expression. BMC Genom. 2010, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Rahmani, M.; Dent, P.; Grant, S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell Biol. 2005, 25, 5429–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlknecht, U.; Voelter-Mahlknecht, S. Chromosomal characterization and localization of the NAD+-dependent histone deacetylase gene sirtuin 1 in the mouse. Int. J. Mol. Med. 2009, 23, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Voelter-Mahlknecht, S.; Mahlknecht, U. Cloning, chromosomal characterization and mapping of the NAD-dependent histone deacetylases gene sirtuin 1. Int. J. Mol. Med. 2006, 17, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, K.; Chen, X.; Meng, H.; Song, M.; Wang, Y.; Xu, X.; Bai, Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol. Biol. 2012, 13, 4. [Google Scholar] [CrossRef] [Green Version]
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menéndez, A.; García-Ruiz, C.; Sancho-Bru, P.; Marí, M.; Caballeria, J.; Rothlin, C.V.; et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678. [Google Scholar] [PubMed] [Green Version]
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 349–368. [Google Scholar] [PubMed]
- McCubbrey, A.L.; Nelson, J.D.; Stolberg, V.R.; Blakely, P.K.; McCloskey, L.; Janssen, W.J.; Freeman, C.M.; Curtis, J.L. MicroRNA-34a Negatively Regulates Efferocytosis by Tissue Macrophages in Part via SIRT1. J. Immunol. 2016, 196, 1366–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24, 3145–3159. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Shentu, T.P.; Wen, L.; Johnson, D.; Shyy, J. Regulation of SIRT1 by Oxidative Stress-Responsive miRNAs and A Systematic Approach to Identify Its Role in the Endothelium. Antioxid. Redox Signal. 2013, 19, 1522–1538. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zhao, Y.; Wu, X.; Xia, M.; Fang, M.; Iwasaki, Y.; Sha, J.; Chen, Q.; Xu, Y.; Shen, A. Interferon gamma (IFN-γ) disrupts energy expenditure and metabolic homeostasis by suppressing SIRT1 transcription. Nucleic. Acids Res. 2012, 40, 1609–1620. [Google Scholar] [CrossRef]
- Anrather, J.; Racchumi, G.; Iadecola, C. NF-κB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J. Biol. Chem. 2006, 281, 5657–5667. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Dorman, L.; Viggiano, V.; Cippitelli, M.; Ghosh, P.; Rice, N.; Young, H.A. Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. J. Biol. Chem. 1997, 272, 30412–30420. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hu, J.; Wang, X.; Zhao, X.; Li, Z.; Niu, J.; Steer, C.J.; Zheng, G.; Song, G. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-κB-TNFα pathway. J. Hepatol. 2019, 70, 87–96. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, C.R. Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 2017, 67, 1104–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S. Hepatic polyploidy and liver growth control. Semin. Cancer Biol. 2000, 10, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Serasanambati, M.; Chilakapati, S.R. Function of Nuclear Factor Kappa B (NF-kB) in human diseases—A Review. S. Indian J. Biol. Sci. 2016, 2, 368. [Google Scholar] [CrossRef]
- Wullaert, A.; van Loo, G.; Heyninck, K.; Beyaert, R. Hepatic Tumor Necrosis Factor Signaling and Nuclear Factor-κB: Effects on Liver Homeostasis and Beyond. Endocr. Rev. 2007, 28, 365–386. [Google Scholar] [CrossRef]
- Shuh, M.; Bohorquez, H.; Loss, G.E., Jr.; Cohen, A.J. Tumor Necrosis Factor-α: Life and Death of Hepatocytes During Liver Ischemia/Reperfusion Injury. Ochsner. J. 2013, 13, 119–130. [Google Scholar]
- Elsharkawy, A.M.; Mann, D.A. Nuclear factor-κB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef]
- Beg, A.A.; Baltimore, D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996, 274, 782–784. [Google Scholar]
- Doi, T.S.; Marino, M.W.; Takahashi, T.; Yoshida, T.; Sakakura, T.; Old, L.J.; Obata, Y. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl. Acad. Sci. USA 1999, 96, 2994–2999. [Google Scholar]
- Czauderna, C.; Castven, D.; Mahn, F.; Marquardt, J. Context-Dependent Role of NF-κB Signaling in Primary Liver Cancer—From Tumor Development to Therapeutic Implications. Cancers 2019, 11, 1053. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Assmus, U.; Wustefeld, T.; Meyer zu Vilsendorf, A.; Roskams, T.; Schmidt-Supprian, M.; Rajewsky, K.; Brenner, D.A.; Manns, M.P.; Pasparakis, M.; et al. Deletion of IKK2 in hepatocytes does not sensitize these cells to TNF-induced apoptosis but protects from ischemia/reperfusion injury. J. Clin. Investig. 2005, 115, 849–859. [Google Scholar] [PubMed]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. Mechanisms of liver disease: Cross-talk between the NF-kappaB and JNK pathways. Biol. Chem. 2009, 390, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.R.; Lee, S.H.; Jung, G. The hepatitis B viral X protein activates NF-κB signaling pathway through the up-regulation of TBK1. FEBS Lett. 2010, 584, 525–530. [Google Scholar] [PubMed]
- Dhamija, E.; Paul, S.B.; Kedia, S. Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern. Indian J. Med. Res. 2019, 149, 9–17. [Google Scholar] [PubMed]
- Xu, W.; Yu, J. Chapter 20—Obesity and Hepatocellular Carcinoma. In Muriel PBT-LP; Muriel, P., Ed.; Academic Press: Boston, UK, 2017; pp. 267–277. [Google Scholar]
- Li, X.; Wang, X.; Gao, P. Diabetes Mellitus and Risk of Hepatocellular Carcinoma. BioMed Res. Int. 2017, 2017, 5202684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Tang, W.J.; Yin, J.J.; Zhou, B.J. Signal transductions and nonalcoholic fatty liver: A mini-review. Int. J. Clin. Exp. Med. 2014, 7, 1624–1631. [Google Scholar]
- Carlsen, H.; Haugen, F.; Zadelaar, S.; Kleemann, R.; Kooistra, T.; Drevon, C.A.; Blomhoff, R. Diet-induced obesity increases NF-kappaB signaling in reporter mice. Genes Nutr. 2009, 4, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Santani, D. Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol. Rep. 2009, 61, 595–603. [Google Scholar] [CrossRef]
- Sun, B.; Karin, M. NF-κB signaling, liver disease and hepatoprotective agents. Oncogene 2008, 27, 6228–6244. [Google Scholar] [CrossRef] [Green Version]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer cells in the liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar]
- Zeng, T.; Zhang, C.-L.; Xiao, M.; Yang, R.; Xie, K.-Q. Critical Roles of Kupffer Cells in the Pathogenesis of Alcoholic Liver Disease: From Basic Science to Clinical Trials. Front. Immunol. 2016, 7, 538. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Tacke, F. Macrophages in obesity and non-alcoholic fatty liver disease: Crosstalk with metabolism. JHEP Rep. 2019, 1, 30–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2019, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer cells in the pathogenesis of liver disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Son, G.; Iimuro, Y.; Seki, E.; Hirano, T.; Kaneda, Y.; Fujimoto, J. Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am. J. Physiol. Gastrointest Liver Physiol. 2007, 293, 631–639. [Google Scholar] [CrossRef]
- Soares, J.-B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [Green Version]
- Louis, H.; Le Moine, O.; Goldman, M.; Devière, J. Modulation of liver injury by interleukin-10. Acta Gastroenterol. Belg. 2003, 66, 7–14. [Google Scholar]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Li, J.-T.; Liao, Z.-X.; Ping, J.; Xu, D.; Wang, H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J. Gastroenterol. 2008, 43, 419–428. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Ying, H.-Z.; Chen, Q.; Zhang, W.-Y.; Zhang, H.-H.; Ma, Y.; Zhang, S.-Z.; Fang, J.; Yu, C.-H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.; Vogel, A. The Platelet-derived growth factor. Cell 1978, 14, 203–210. [Google Scholar] [CrossRef]
- De Oliveira da Silva, B.; Ramos, L.F.; Moraes, K.C.M. Molecular interplays in hepatic stellate cells: Apoptosis, senescence, and phenotype reversion as cellular connections that modulate liver fibrosis. Cell Biol. Int. 2017, 41, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anan, A.; Baskin-Bey, E.S.; Bronk, S.F.; Werneburg, N.W.; Shah, V.H.; Gores, G.J. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology 2006, 43, 335–344. [Google Scholar] [CrossRef]
- Yang, H.; Feldser, H.G.; Zhang, W.; Zhong, Z.; Loh, C.; Ellis, J. SIRT1 activators promote p65 deacetylation and suppress TNFa stimulated NF-kB activation. FASEB J. 2011, 25, 945-12. [Google Scholar]
- Yang, S.-R.; Wright, J.; Bauter, M.; Seweryniak, K.; Kode, A.; Rahman, I. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-κB in macrophages in vitro and in rat lungs in vivo: Implications for chronic inflammation and aging. Am. J. Physiol. Cell Mol. Physiol. 2007, 292, L567–L576. [Google Scholar] [CrossRef]
- Gao, R.; Chen, J.; Hu, Y.; Li, Z.; Wang, S.; Shetty, S.; Fu, J. Sirt1 deletion leads to enhanced inflammation and aggravates endotoxin-induced acute kidney injury. PLoS ONE 2014, 9, e98909. [Google Scholar]
- Revollo, J.R.; Li, X. The ways and means 6 that fine tune Sirt1 activity. Trends Biochem. Sci. 2013, 38, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.-X.; Zhang, R.; Huang, L.; Zhu, L.-H.; Jiang, D.-S.; Chen, H.-Z.; Zhang, Y.; Tian, S.; Zhang, X.-F.; Zhang, X.-D.; et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J. Hepatol. 2015, 62, 111–120. [Google Scholar] [PubMed]
- Wang, X.-A.; Zhang, R.; Jiang, D.; Deng, W.; Zhang, S.; Deng, S.; Zhong, J.; Wang, T.; Zhu, L.-H.; Yang, L.; et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology 2013, 58, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Kalliora, C.; Kyriazis, I.D.; Oka, S.; Lieu, M.J.; Yue, Y.; Area-Gomez, E.; Pol, C.J.; Tian, Y.; Mizushima, W.; Chin, A.; et al. Dual PPARα/γ activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. JCI Insight 2019, 4, e129556. [Google Scholar]
- Johnson, S.; Imai, S.-I. NAD (+) biosynthesis, aging, and disease. F1000 Res. 2018, 7, 132. [Google Scholar]
- Imai, S.-I. Nicotinamide phosphoribosyltransferase (Nampt): A link between NAD biology, metabolism, and diseases. Curr. Pharm. Des. 2009, 15, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-F.; Wang, X.-N.; Huang, C.-C.; Hu, L.; Xiao, Y.-F.; Guan, X.-H.; Qian, Y.-S.; Deng, K.-Y.; Xin, H.-B. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKalpha/SREBP1 signaling pathway. Lipids Health Dis. 2017, 16, 82. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar]
- Caton, P.W.; Kieswich, J.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Nicotinamide mononucleotide protects against pro-inflammatory cytokine-mediated impairment of mouse islet function. Diabetologia 2011, 54, 3083–3092. [Google Scholar]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreuz, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Yang, S.J. Nicotinamide riboside regulates inflammation and mitochondrial markers in AML12 hepatocytes. Nutr. Res. Pract. 2019, 13, 3–10. [Google Scholar] [PubMed]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Wakade, C.; Giri, B.; Malik, A.; Khodadadi, H.; Morgan, J.C.; Chong, R.K.; Baban, B. Niacin modulates macrophage polarization in Parkinson’s disease. J. Neuroimmunol. 2018, 320, 76–79. [Google Scholar] [CrossRef]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar]
- Ding, Y.; Li, Y.; Wen, A. Effect of niacin on lipids and glucose in patients with type 2 diabetes: A meta-analysis of randomized, controlled clinical trials. Clin. Nutr. 2015, 34, 838–844. [Google Scholar]
- Kashyap, M.L.; Ganji, S.; Nakra, N.K.; Kamanna, V.S. Niacin for treatment of nonalcoholic fatty liver disease (NAFLD): Novel use for an old drug? J. Clin. Lipidol. 2019, 13, 873–879. [Google Scholar]
- Kwon, H.-S.; Ott, M. The ups and downs of SIRT1. Trends Biochem. Sci. 2008, 33, 517–525. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Pullmann, R.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 Regulates SIRT1 Expression. Mol. Cell. 2007, 25, 543–557. [Google Scholar] [CrossRef] [Green Version]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. Landmark Ed. 2012, 17, 189–205. [Google Scholar] [PubMed] [Green Version]
- Wang, J.; Kainrad, N.; Shen, H.; Zhou, Z.; Rote, P.; Zhang, Y.; Nagy, L.; Wu, J.; You, M. Hepatic Knockdown of Splicing Regulator Slu7 Ameliorates Inflammation and Attenuates Liver Injury in Ethanol-Fed Mice. Am. J. Pathol. 2018, 188, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 Protein Kinases: Conserved Guardians of Cellular Energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [PubMed]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.W.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Görgün, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.J.; Zhang, Z.; Jung, D.Y.; Jun, J.Y.; Ma, Z.; Jones, K.E.; Chan, S.Y.; Kim, J.K. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 2009, 58, 2536–2546. [Google Scholar] [CrossRef] [Green Version]
- Carroll, K.C.; Viollet, B.; Suttles, J. AMPKalpha1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 2013, 94, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell. 2008, 14, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Chyau, C.-C.; Wang, H.-F.; Zhang, W.-J.; Chen, C.-C.; Huang, S.-H.; Chang, C.-C.; Peng, R.Y. Antrodan Alleviates High-Fat and High-Fructose Diet-Induced Fatty Liver Disease in C57BL/6 Mice Model via AMPK/Sirt1/SREBP-1c/PPARγ Pathway. Int. J. Mol. Sci. 2020, 21, 360. [Google Scholar] [CrossRef] [Green Version]
- Nagappan, A.; Kim, J.-H.; Jung, Y.D.; Jung, H.M. Cryptotanshinone from the Salvia miltiorrhiza Bunge Attenuates Ethanol-Induced Liver Injury by Activation of AMPK/SIRT1 and Nrf2 Signaling Pathways. Int. J. Mol. Sci. 2019, 21, 265. [Google Scholar] [CrossRef] [Green Version]
- Luna, A.; Aladjem, M.I.; Kohn, K.W. SIRT1/PARP1 crosstalk: Connecting DNA damage and metabolism. Genome Integr. 2013, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: Molecular Mechanisms and Beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.T.; Joe, Y. Antagonistic crosstalk between SIRT1, PARP-1, and -2 in the regulation of chronic inflammation associated with aging and metabolic diseases. Integr. Med. Res. 2014, 3, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Hogan-Cann, A.D.; Kamboj, A.; Roy Chowdhury, S.K.; Aghanoori, M.-R.; Fernyhough, P.; Anderson, C.M. Poly(ADP-ribose) polymerase-1 inhibits mitochondrial respiration by suppressing PGC-1alpha activity in neurons. Neuropharmacology 2019, 160, 107755. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.; Daitoku, H.; Yoshimochi, K.; Miwa, M.; Fukamizu, A. Regulation of FOXO1-mediated transcription and cell proliferation by PARP-1. Biochem. Biophys. Res. Commun. 2009, 382, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 Activation Mimics Low Energy Levels and Protects against Diet-Induced Metabolic Disorders by Enhancing Fat Oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. [Google Scholar] [CrossRef]
- Henning, R.J.; Bourgeois, M.; Harbison, R.D. Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders. Cardiovasc. Toxicol. 2018, 18, 493–506. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C. The Role of PARP-1 and PARP-2 Enzymes in Metabolic Regulation and Disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [PubMed] [Green Version]
- Masutani, M.; Nakagama, H.; Sugimura, T. Poly-ADP-ribosylation in health and disease. Cell Mol. Life Sci. C 2005, 62, 769–783. [Google Scholar]
- Mukhopadhyay, P.; Rajesh, M.; Cao, Z.; Horváth, B.; Park, O.; Wang, H.; Liaudet, L.; Hamdaoui, N.; Lafdil, F.; Haskó, G.; et al. Poly (ADP-ribose) polymerase-1 is a key mediator of liver inflammation and fibrosis. Hepatology 2014, 59, 1998–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gariani, K.; Ryu, D.; Menzies, K.J.; Yi, H.-S.; Stein, S.; Zhang, H.; Perino, A.; Lemos, V.; Katsyuba, E.; Jha, P.; et al. Inhibiting poly ADP-ribosylation increases fatty acid oxidation and protects against fatty liver disease. J. Hepatol. 2017, 66, 132–141. [Google Scholar]
- Rusin, S.F.; Adamo, M.E.; Kettenbach, A.N. Identification of Candidate Casein Kinase 2 Substrates in Mitosis by Quantitative Phosphoproteomics. Front. Cell Dev. Biol. 2017, 5, 97. [Google Scholar]
- Gibson, S.A.; Benveniste, E.N. Protein Kinase CK2: An Emerging Regulator of Immunity. Trends Immunol. 2018, 39, 82–85. [Google Scholar] [CrossRef]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: Linking development and cancer. Cell Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef]
- Singh, N.N.; Ramji, D.P. Protein kinase CK2, an important regulator of the inflammatory response? J. Mol. Med. 2008, 86, 887. [Google Scholar] [CrossRef]
- Roelants, C.; Giacosa, S.; Duchemin-Pelletier, E.; McLeer-Florin, A.; Tisseyre, C.; Aubert, C.; Champelovier, P.; Boutonnat, J.; Descotes, J.L.; Rambeaud, J.J.; et al. Dysregulated Expression of Protein Kinase CK2 in Renal Cancer; Pinna, L.A., Ahmed, K., Issinger, O.-G., Szyszka, R., Eds.; Springer International Publishing: Basel, Switzerland, 2015; pp. 241–257. [Google Scholar]
- Choi, S.E.; Kwon, S.; Seok, S.; Xiao, Z.; Lee, K.-W.; Kang, Y.; Li, X.; Shinoda, K.; Kajimura, S.; Kemper, B.; et al. Obesity-Linked Phosphorylation of SIRT1 by Casein Kinase 2 Inhibits Its Nuclear Localization and Promotes Fatty Liver. Mol. Cell Biol. 2017, 37, e00006–e00017. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.-N.; Dong, L.-H.; Li, H.; Pei, Q.-Q.; Miao, S.-B.; Zhang, F.; Zhang, D.-D.; Chen, R.; Yin, Y.-J.; Lin, Y.-L.; et al. CKII-SIRT1-SM22α loop evokes a self-limited inflammatory response in vascular smooth muscle cells. Cardiovasc. Res. 2017, 113, 1198–1207. [Google Scholar] [CrossRef]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [PubMed]
- Oberle, C.; Huai, J.; Reinheckel, T.; Tacke, M.; Rassner, M.; Ekert, P.G.; Buellesbach, J.; Borner, C. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010, 17, 1167. [Google Scholar] [PubMed] [Green Version]
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Ollinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xavier, S.; Moskowitz-Kassai, E.; Chen, R.; Lu, C.Y.; Sanduski, K.; Špes, A.; Turk, B.; Goligorsky, M.S. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am. J. Pathol. 2012, 180, 973–983. [Google Scholar] [PubMed] [Green Version]
- Oppenheimer, H.; Gabay, O.; Meir, H.; Haze, A.; Kandel, L.; Liebergall, M.; Gagarina, V.; Lee, E.J.; Dvir-Ginzberg, M. 75-kd sirtuin 1 blocks tumor necrosis factor α-mediated apoptosis in human osteoarthritic chondrocytes. Arthritis Rheum. 2012, 64, 718–728. [Google Scholar]
- Werneburg, N.W.; Guicciardi, M.E.; Bronk, S.F.; Gores, G.J. Tumor necrosis factor-α-associated lysosomal permeabilization is cathepsin B dependent. Am. J. Physiol. Liver Physiol. 2002, 283, G947–G956. [Google Scholar]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-α–mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology 2009, 49, 1297–1307. [Google Scholar]
- Moles, A.; Tarrats, N.; Morales, A.; Domínguez, M.; Bataller, R.; Caballería, J.; García-Ruiz, C.; Fernández-Checa, J.C.; Marí, M. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. Am. J. Pathol. 2010, 177, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [PubMed]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschöp, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [PubMed] [Green Version]
- Ponugoti, B.; Kim, D.-H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef] [Green Version]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Chimento, A.; De Amicis, F.; Sirianni, R.; Sinicropi, M.S.; Puoci, F.; Casaburi, I.; Saturnino, C.; Pezzi, V. Progress to Improve Oral Bioavailability and Beneficial Effects of Resveratrol. Int. J. Mol. Sci. 2019, 20, 1381. [Google Scholar] [CrossRef] [Green Version]
- Pirola, L.; Frojdo, S. Resveratrol: One molecule, many targets. IUBMB Life 2008, 60, 323–332. [Google Scholar]
- Guarente, L.; Baur, J.; Mai, A. Revelations into resveratrol’s mechanism. Nat. Med. 2012, 18, 500–501. [Google Scholar]
- Venkatasubramanian, S.; Noh, R.M.; Daga, S.; Langrish, J.P.; Joshi, N.V.; Mills, N.L.; Hoffmann, E.; Jacobson, E.W.; Vlasuk, G.P.; Waterhouse, B.R.; et al. Cardiovascular Effects of a Novel SIRT1 Activator, SRT2104, in Otherwise Healthy Cigarette Smokers. J. Am. Heart Assoc. 2020, 2, e000042. [Google Scholar] [CrossRef] [Green Version]
- Meer, A.; Scicluna, B.; Moerland, P.; Lin, J.; Jacobson, E.; Vlasuk, G.; van der Poll, T. The Selective Sirtuin 1 Activator SRT2104 Reduces Endotoxin-Induced Cytokine Release and Coagulation Activation in Humans. Crit. Care Med. 2015, 43, e199–e202. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [PubMed] [Green Version]
- Musso, G.; Gambino, R.; Bo, S.; Uberti, B.; Biroli, G.; Pagano, G.; Cassader, M. Should nonalcoholic fatty liver disease be included in the definition of metabolic syndrome? A cross-sectional comparison with Adult Treatment Panel III criteria in nonobese nondiabetic subjects. Diabetes Care 2008, 31, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, A.; Conti, V.; Russomanno, G.; Dallio, M.; Masarone, M.; Stiuso, P.; Tuccillo, C.; Caraglia, M.; Manzo, V.; Persico, M.; et al. A Long-term Treatment with Silybin in Patients with Non-alcoholic Steatohepatitis Stimulates Catalase Activity in Human Endothelial Cells. In Vivo 2017, 31, 609–618. [Google Scholar] [PubMed] [Green Version]
- Conti, V.; Corbi, G.; Simeon, V.; Russomanno, G.; Manzo, V.; Ferrara, N.; Filippelli, A. Aging-related changes in oxidative stress response of human endothelial cells. Aging Clin. Exp. Res. 2015, 27, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [PubMed]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Gonçalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [PubMed]
- Farghali, H.; Kemelo, M.; Kutinová-Canová, N. SIRT1 Modulators in Experimentally Induced Liver Injury. Oxid. Med. Cell Longev. 2019, 2019, 1–15. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858. https://doi.org/10.3390/ijms21113858
de Gregorio E, Colell A, Morales A, Marí M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. International Journal of Molecular Sciences. 2020; 21(11):3858. https://doi.org/10.3390/ijms21113858
Chicago/Turabian Stylede Gregorio, Estefanía, Anna Colell, Albert Morales, and Montserrat Marí. 2020. "Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease" International Journal of Molecular Sciences 21, no. 11: 3858. https://doi.org/10.3390/ijms21113858
APA Stylede Gregorio, E., Colell, A., Morales, A., & Marí, M. (2020). Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. International Journal of Molecular Sciences, 21(11), 3858. https://doi.org/10.3390/ijms21113858