Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes

Abstract
:1. Introduction
2. SIRT3 Regulates Mitochondrial Metabolism and Insulin Sensitivity
2.1. SIRT3 in Mitochondrial Function
2.2. SIRT3 in Redox Homeostasis
2.3. SIRT3 in Glucose Uptake, Insulin Sensitivity and Fatty Acid Metabolism
2.4. SIRT3 in Insulin Secretion
2.5. SIRT3 in the Differentiation of Myocytes and Adipocytes
3. SIRT4 Regulates Mitochondrial Metabolism and Insulin Sensitivity
3.1. SIRT4 in Mitochondrial Function and Redox Homeostasis
3.2. SIRT4 in Insulin Secretion
3.3. SIRT4 in Fatty Acid Oxidation
4. SIRT5 Regulates Mitochondrial Metabolism and Insulin Sensitivity
4.1. SIRT5 in Mitochondrial Function and Redox Homeostasis
4.2. SIRT5 in Glycolysis, Fatty Acid Oxidation and Ammonia Metabolism
4.3. SIRT5 in BAT Function and Adipogenic Differentiation
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rother, K.I. Diabetes treatment—bridging the divide. N. Engl. J. Med. 2007, 356, 1499–1501. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Lee, J.I.; Suh, Y.H.; Kim, W.; Song, J.H.; Jung, M.H. Mitochondrial dysfunction induces aberrant insulin signalling and glucose utilisation in murine C2C12 myotube cells. Diabetologia 2006, 49, 1924–1936. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wang, C.C.; Huang, H.C.; Wei, Y.H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Franquesa, A.; Patti, M.E. Insulin resistance and mitochondrial dysfunction. Adv. Exp. Med. Biol. 2017, 982, 465–520. [Google Scholar]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The mitochondrial acylome emerges: Proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Jiang, X.; Ma, H.; Wang, Y.; Xue, P.; Liu, Y. SIRT1 and insulin resistance. J. Diabetes Complicat. 2016, 30, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Lombard, D.B. Functions of the sirtuin deacylase SIRT5 in normal physiology and pathobiology. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 311–334. [Google Scholar] [CrossRef]
- Cortes-Rojo, C.; Vargas-Vargas, M.A.; Olmos-Orizaba, B.E.; Rodriguez-Orozco, A.R.; Calderon-Cortes, E. Interplay between NADH oxidation by complex I, glutathione redox state and sirtuin-3, and its role in the development of insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165801–165817. [Google Scholar] [CrossRef]
- Min, Z.; Gao, J.; Yu, Y. The roles of mitochondrial SIRT4 in cellular metabolism. Front. Endocrinol. (Lausanne) 2018, 9, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Wu, S.B.; Wei, Y.H. Roles of sirtuins in the regulation of antioxidant defense and bioenergetic function of mitochondria under oxidative stress. Free Radic. Res. 2014, 48, 1070–1084. [Google Scholar] [CrossRef]
- Cho, E.H. SIRT3 as a regulator of non-alcoholic fatty liver disease. J. Lifestyle Med. 2014, 4, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [Green Version]
- Cimen, H.; Han, M.J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry 2010, 49, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.T.; Lee, H.C.; Liao, C.C.; Wei, Y.H. Regulation of mitochondrial FoF1ATPase activity by Sirt3-catalyzed deacetylation and its deficiency in human cells harboring 4977 bp deletion of mitochondrial DNA. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, E.; O’Neill, B.T.; Rardin, M.J.; Kleinridders, A.; Ilkeyeva, O.R.; Ussar, S.; Bain, J.R.; Lee, K.Y.; Verdin, E.M.; Newgard, C.B.; et al. Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 2013, 62, 3404–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwer, B.; Bunkenborg, J.; Verdin, R.O.; Andersen, J.S.; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 2006, 103, 10224–10229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany N. Y.) 2010, 2, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Gao, B.; Li, N.; Wang, J.; Qiu, C.; Zhang, G.; Liu, M.; Zhang, R.; Li, C.; Ji, G.; et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1973–1983. [Google Scholar] [CrossRef]
- Drago, I.; Pizzo, P.; Pozzan, T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011, 30, 4119–4125. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.H.; Wei, Y.H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J. Biomed. Sci. 2017, 24, 70. [Google Scholar] [CrossRef]
- Draznin, B.; Sussman, K.E.; Eckel, R.H.; Kao, M.; Yost, T.; Sherman, N.A. Possible role of cytosolic free calcium concentrations in mediating insulin resistance of obesity and hyperinsulinemia. J. Clin. Invest. 1988, 82, 1848–1852. [Google Scholar] [CrossRef]
- Draznin, B.; Lewis, D.; Houlder, N.; Sherman, N.; Adamo, M.; Garvey, W.T.; LeRoith, D.; Sussman, K. Mechanism of insulin resistance induced by sustained levels of cytosolic free calcium in rat adipocytes. Endocrinology 1989, 125, 2341–2349. [Google Scholar] [CrossRef]
- Dai, S.H.; Chen, T.; Wang, Y.H.; Zhu, J.; Luo, P.; Rao, W.; Yang, Y.F.; Fei, Z.; Jiang, X.F. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int. J. Mol. Sci. 2014, 15, 14591–14609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Jiang, Y.; Wu, H.; Sun, F.; Li, Y.; He, H.; Wang, B.; Lu, Z.; Hu, Y.; Wei, X.; et al. Inhibition of mitochondrial calcium overload by SIRT3 prevents obesity- or age-related whitening of brown adipose tissue. Diabetes 2020, 69, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ren, X.; Gowda, A.S.; Shan, Y.; Zhang, L.; Yuan, Y.S.; Patel, R.; Wu, H.; Huber-Keener, K.; Yang, J.W.; et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 2013, 4, e731–e741. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Kitada, M.; Monno, I.; Kanasaki, K.; Watanabe, A.; Koya, D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox Rep. 2018, 23, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlicker, C.; Gertz, M.; Papatheodorou, P.; Kachholz, B.; Becker, C.F.; Steegborn, C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008, 382, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lantier, L.; Williams, A.S.; Williams, I.M.; Yang, K.K.; Bracy, D.P.; Goelzer, M.; James, F.D.; Gius, D.; Wasserman, D.H. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes 2015, 64, 3081–3092. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Bharathi, S.S.; Zhang, Y.; Mohsen, A.W.; Uppala, R.; Balasubramani, M.; Schreiber, E.; Uechi, G.; Beck, M.E.; Rardin, M.J.; Vockley, J.; et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem. 2013, 288, 33837–33847. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295–e23300. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.H.; Cypess, A.M.; Kahn, C.R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug Discov. 2010, 9, 465–482. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef]
- Zhou, Y.; Chung, A.C.K.; Fan, R.; Lee, H.M.; Xu, G.; Tomlinson, B.; Chan, J.C.N.; Kong, A.P.S. Sirt3 deficiency increased the vulnerability of pancreatic beta cells to oxidative stress-induced dysfunction. Antioxid. Redox Signal. 2017, 27, 962–976. [Google Scholar] [CrossRef]
- Caton, P.W.; Richardson, S.J.; Kieswich, J.; Bugliani, M.; Holland, M.L.; Marchetti, P.; Morgan, N.G.; Yaqoob, M.M.; Holness, M.J.; Sugden, M.C. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 2013, 56, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Lee, J.S.; Oh, J.E.; Nan, J.; Lee, H.; Jung, H.S.; Chung, S.S.; Park, K.S. SIRT3 overexpression attenuates palmitate-induced pancreatic beta-cell dysfunction. PLoS ONE 2015, 10, e0124744–e0124756. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Wu, Y.T.; Tsai, C.L.; Wei, Y.H. Current understanding and future perspectives of the roles of sirtuins in the reprogramming and differentiation of pluripotent stem cells. Exp. Biol. Med. (Maywood) 2018, 243, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Denu, R.A. SIRT3 enhances mesenchymal stem cell longevity and differentiation. Oxid. Med. Cell. Longev. 2017, 2017, 5841716–5841726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel Khalek, W.; Cortade, F.; Ollendorff, V.; Lapasset, L.; Tintignac, L.; Chabi, B.; Wrutniak-Cabello, C. SIRT3, a mitochondrial NAD+-dependent deacetylase, is involved in the regulation of myoblast differentiation. PLoS ONE 2014, 9, e114388–e114407. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.T.; Chi, K.T.; Lan, Y.W.; Chan, J.C.; Ma, Y.S.; Wei, Y.H. Depletion of Sirt3 leads to the impairment of adipogenic differentiation and insulin resistance via interfering mitochondrial function of adipose-derived human mesenchymal stem cells. Free Radic Res. 2018, 52, 1398–1415. [Google Scholar] [CrossRef]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Diaz-Delfin, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.; Titus, A.S.; Banerjee, K.K.; George, S.; Lin, W.; Deota, S.; Saha, A.K.; Nakamura, K.; Gut, P.; Verdin, E.; et al. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging (Albany N. Y.) 2013, 5, 835–849. [Google Scholar] [CrossRef] [Green Version]
- Nasrin, N.; Wu, X.; Fortier, E.; Feng, Y.; Bare, O.C.; Chen, S.; Ren, X.; Wu, Z.; Streeper, R.S.; Bordone, L. SIRT4 regulates fatty acid oxidation and mitochondrial gene expression in liver and muscle cells. J. Biol. Chem. 2010, 285, 31995–32002. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Jiang, M.; Wu, X.; Diao, F.; Qiu, D.; Hou, X.; Wang, H.; Li, L.; Li, C.; Ge, J.; et al. SIRT4 is essential for metabolic control and meiotic structure during mouse oocyte maturation. Aging Cell 2018, 17, e12789–e12798. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.X.; Tang, X.; An, X.Z.; Xie, X.M.; Chen, X.F.; Zhao, X.; Hao, D.L.; Chen, H.Z.; Liu, D.P. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur. Heart J. 2017, 38, 1389–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.; Anand, R.; Altinoluk-Hambuchen, S.; Ezzahoini, H.; Stefanski, A.; Iram, A.; Bergmann, L.; Urbach, J.; Bohler, P.; Hansel, J.; et al. SIRT4 interacts with OPA1 and regulates mitochondrial quality control and mitophagy. Aging (Albany N. Y.) 2017, 9, 2163–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007, 282, 33583–33592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.A.; Huynh, F.K.; Fisher-Wellman, K.; Stuart, J.D.; Peterson, B.S.; Douros, J.D.; Wagner, G.R.; Thompson, J.W.; Madsen, A.S.; Green, M.F.; et al. SIRT4 is a lysine deacylase that controls leucine metabolism and insulin secretion. Cell Metab. 2017, 25, 838–855. [Google Scholar] [CrossRef] [Green Version]
- Zaganjor, E.; Vyas, S.; Haigis, M.C. SIRT4 is a regulator of insulin secretion. Cell Chem. Biol. 2017, 24, 656–658. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.R.; Fang, S.R.; Fu, Y.C.; Zhou, X.H.; Xu, M.Y.; Xu, W.C. Calorie restriction on insulin resistance and expression of SIRT1 and SIRT4 in rats. Biochem. Cell Biol. 2010, 88, 715–722. [Google Scholar] [CrossRef]
- de Pinho, L.; Andrade, J.M.; Paraiso, A.; Filho, A.B.; Feltenberger, J.D.; Guimaraes, A.L.; de Paula, A.M.; Caldeira, A.P.; de Carvalho Botelho, A.C.; Campagnole-Santos, M.J.; et al. Diet composition modulates expression of sirtuins and renin-angiotensin system components in adipose tissue. Obesity (Silver Spring) 2013, 21, 1830–1835. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar] [PubMed]
- Laurent, G.; German, N.J.; Saha, A.K.; de Boer, V.C.; Davies, M.; Koves, T.R.; Dephoure, N.; Fischer, F.; Boanca, G.; Vaitheesvaran, B.; et al. SIRT4 coordinates the balance between lipid synthesis and catabolism by repressing malonyl CoA decarboxylase. Mol. Cell 2013, 50, 686–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, G.; de Boer, V.C.; Finley, L.W.; Sweeney, M.; Lu, H.; Schug, T.T.; Cen, Y.; Jeong, S.M.; Li, X.; Sauve, A.A.; et al. SIRT4 represses peroxisome proliferator-activated receptor alpha activity to suppress hepatic fat oxidation. Mol. Cell. Biol. 2013, 33, 4552–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Meyer, J.G.; Cai, W.; Softic, S.; Li, M.E.; Verdin, E.; Newgard, C.; Schilling, B.; Kahn, C.R. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol. Cell 2019, 74, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Shuai, L.; Zhang, L.N.; Li, B.H.; Tang, C.L.; Wu, L.Y.; Li, J.; Li, J.Y. SIRT5 regulates brown adipocyte differentiation and browning of subcutaneous white adipose tissue. Diabetes 2019, 68, 1449–1461. [Google Scholar] [CrossRef]
- Yu, J.; Sadhukhan, S.; Noriega, L.G.; Moullan, N.; He, B.; Weiss, R.S.; Lin, H.; Schoonjans, K.; Auwerx, J. Metabolic characterization of a Sirt5 deficient mouse model. Sci. Rep. 2013, 3, 2806–2812. [Google Scholar] [CrossRef]
- Guedouari, H.; Daigle, T.; Scorrano, L.; Hebert-Chatelain, E. Sirtuin 5 protects mitochondria from fragmentation and degradation during starvation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 169–176. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wang, F.; Sun, R.; Chen, X.; Zhang, M.; Xu, Q.; Wang, Y.; Wang, S.; Xiong, Y.; Guan, K.L.; et al. SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 2016, 17, 811–822. [Google Scholar] [CrossRef]
- Lin, Z.F.; Xu, H.B.; Wang, J.Y.; Lin, Q.; Ruan, Z.; Liu, F.B.; Jin, W.; Huang, H.H.; Chen, X. SIRT5 desuccinylates and activates SOD1 to eliminate ROS. Biochem. Biophys. Res. Commun. 2013, 441, 191–195. [Google Scholar] [CrossRef]
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124–e45141. [Google Scholar] [CrossRef] [PubMed]
- Xiangyun, Y.; Xiaomin, N.; Linping, G.; Yunhua, X.; Ziming, L.; Yongfeng, Y.; Zhiwei, C.; Shun, L. Desuccinylation of pyruvate kinase M2 by SIRT5 contributes to antioxidant response and tumor growth. Oncotarget 2017, 8, 6984–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Ning, X.; Yu, C.; Ji, X.; Jin, Y.; McNutt, M.A.; Yin, Y. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019, 10, 170–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jukarainen, S.; Heinonen, S.; Ramo, J.T.; Rinnankoski-Tuikka, R.; Rappou, E.; Tummers, M.; Muniandy, M.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; et al. Obesity is associated with low NAD+/SIRT pathway expression in adipose tissue of BMI-discordant monozygotic twins. J. Clin. Endocrinol. Metab. 2016, 101, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Lomb, D.J.; Haigis, M.C.; Guarente, L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 2009, 137, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Frontini, A.; Cinti, S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 2010, 11, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Townsend, K.L.; Tseng, Y.H. Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 2014, 25, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoliga, J.M.; Baur, J.A.; Hausenblas, H.A. Resveratrol and health—A comprehensive review of human clinical trials. Mol. Nutr. Food Res. 2011, 55, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Henagan, T.M.; Cefalu, W.T.; Ribnicky, D.M.; Noland, R.C.; Dunville, K.; Campbell, W.W.; Stewart, L.K.; Forney, L.A.; Gettys, T.W.; Chang, J.S.; et al. In vivo effects of dietary quercetin and quercetin-rich red onion extract on skeletal muscle mitochondria, metabolism, and insulin sensitivity. Genes Nutr. 2015, 10, 451–462. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Yoshino, J. Adipose tissue NAD+ biology in obesity and insulin resistance: From mechanism to therapy. BioEssays 2017, 39, 1600227–1600235. [Google Scholar] [CrossRef] [Green Version]


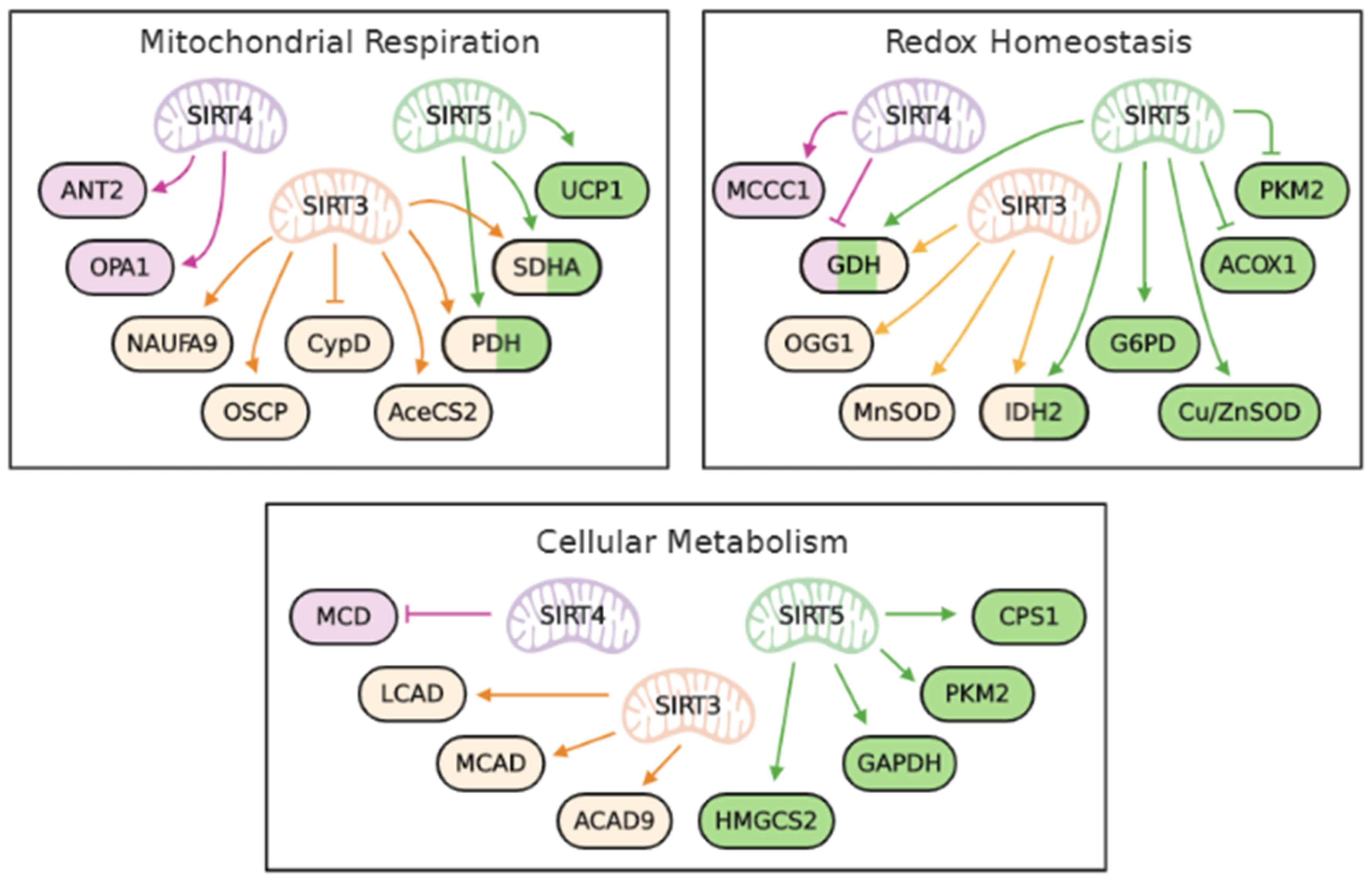{kind=link}
| Functions | Proteins | Activities | Sirtuins | Modifications | Outcomes | References |
|---|---|---|---|---|---|---|
| Mitochondrial respiration &oxidative phosphorylation | NDUFA9 | Activation | SIRT3 | Deacetylation | Increase of Complex I activity | [19] |
| SDHA | Activation | SIRT3 | Deacetylation | Increase of Complex II activity | [20,47,74] | |
| SIRT5 | Desuccinylation | [78] | ||||
| OSCP | Activation | SIRT3 | Deacetylation | Increase of Complex V activity | [21] | |
| ANT2 | Activation | SIRT4 | Deacetylation | Increase of coupled respiration | [61] | |
| UCP1 | Activation | SIRT5 | Desuccinylation | Increase of uncoupled respiration | [74] | |
| Acetyl-CoA availability | PDH | Activation | SIRT3 | Deacetylation | Increase of acetyl-CoA | [22] |
| SIRT5 | Desuccinylation | [78] | ||||
| AceCS2 | Activation | SIRT3 | Deacetylation | Increase of acetyl-CoA | [23,24] | |
| MCD | Inhibition | SIRT4 | Deacetylation | Decrease of acetyl-CoA | [72] | |
| Mitochondrial fusion | OPA1 | Activation | SIRT4 | Deacetylation | Decrease of mitophagy | [64] |
| Mitochondrial permeability transition pore (mPTP) | CypD | Inhibition | SIRT3 | Deacetylation | Inhibition of mPTP opening | [25] |
| PKM2 | Inhibition | SIRT5 | Desuccinylation | Increase of mPTP opening | [83] | |
| Transcriptional regulator | FoxO3A | Activation | SIRT3 | Deacetylation | Increase of adipocyte differentiation | [56] |
| Increase of mitophagy | [26] | |||||
| Increase of antioxidant enzyme level | [39] | |||||
| Antioxidant defense | MnSOD | Activation | SIRT3 | Deacetylation | Decrease of ROS | [36,37,38] |
| Cu/ZnSOD | Activation | SIRT5 | Desuccinylation | Decrease of ROS | [80] | |
| ACOX1 | Inhibition | SIRT5 | Desuccinylation | Decrease of ROS | [81] | |
| NADPH production | GDH | Activation | SIRT3 | Deacetylation | Increase of NADPH | [42] |
| SIRT5 | Desuccinylation | [74,78] | ||||
| Inhibition | SIRT4 | ADP-ribosylation | Decrease of NADPH | [66] | ||
| IDH2 | Activation | SIRT3 | Deacetylation | Increase of NADPH | [40,41] | |
| SIRT5 | Desuccinylation | [78,79] | ||||
| G6PD | Activation | SIRT5 | Deglutarylation | Increase of NADPH | [79] | |
| PKM2 | Inhibition | SIRT5 | Desuccinylation | Increase of NADPH | [82] | |
| MCCC1 | Activation | SIRT4 | Deacetylation | Decrease of NADPH | [67,68] | |
| DNA repair | OGG1 | Activation | SIRT3 | Deacetylation | Increase of DNA repair | [34] |
| Glycolysis | GAPDH | Activation | SIRT5 | Demalonylation | Increase glycolysis | [84] |
| PKM2 | Inhibition | SIRT5 | Desuccinylation | Decrease of glycolysis | [82] | |
| Fatty acid metabolism | LCAD | Activation | SIRT3 | Deacetylation | Increase of fatty acid oxidation | [45,46] |
| MCAD | Activation | SIRT3 | Deacetylation | Increase of fatty acid oxidation | [45,46] | |
| ACAD9 | Activation | SIRT3 | Deacetylation | Increase of fatty acid oxidation | [45,46] | |
| HMGCS2 | Activation | SIRT5 | Desuccinylation | Increase of ketogenesis | [86] | |
| Urea cycle | CPS1 | Activation | SIRT5 | Deacetylation | Detoxification of ammonia | [88] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-H.; Wei, Y.-H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 5266. https://doi.org/10.3390/ijms21155266
Wang C-H, Wei Y-H. Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. International Journal of Molecular Sciences. 2020; 21(15):5266. https://doi.org/10.3390/ijms21155266
Chicago/Turabian StyleWang, Chih-Hao, and Yau-Huei Wei. 2020. "Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes" International Journal of Molecular Sciences 21, no. 15: 5266. https://doi.org/10.3390/ijms21155266
APA StyleWang, C. -H., & Wei, Y. -H. (2020). Roles of Mitochondrial Sirtuins in Mitochondrial Function, Redox Homeostasis, Insulin Resistance and Type 2 Diabetes. International Journal of Molecular Sciences, 21(15), 5266. https://doi.org/10.3390/ijms21155266