Albendazole-Induced SIRT3 Upregulation Protects Human Leukemia K562 Cells from the Cytotoxicity of MCL1 Suppression


Abstract
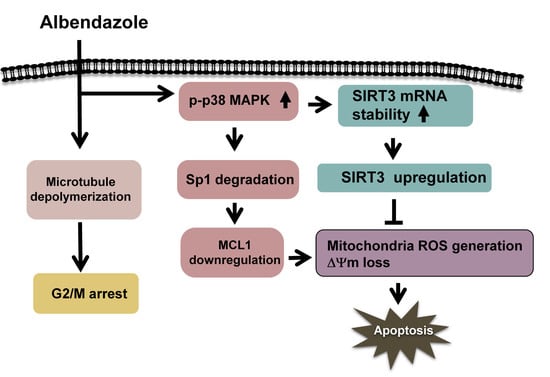
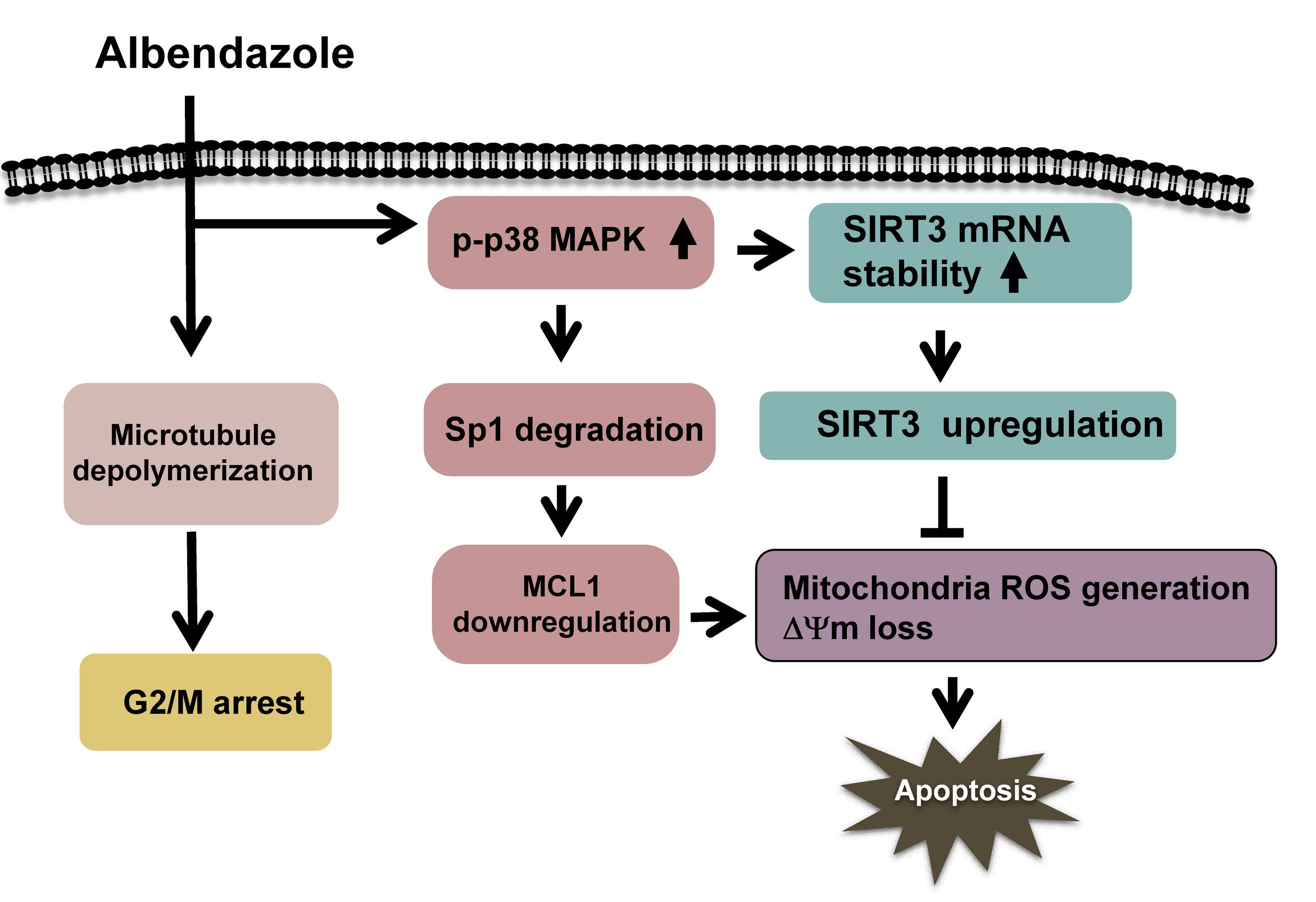
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
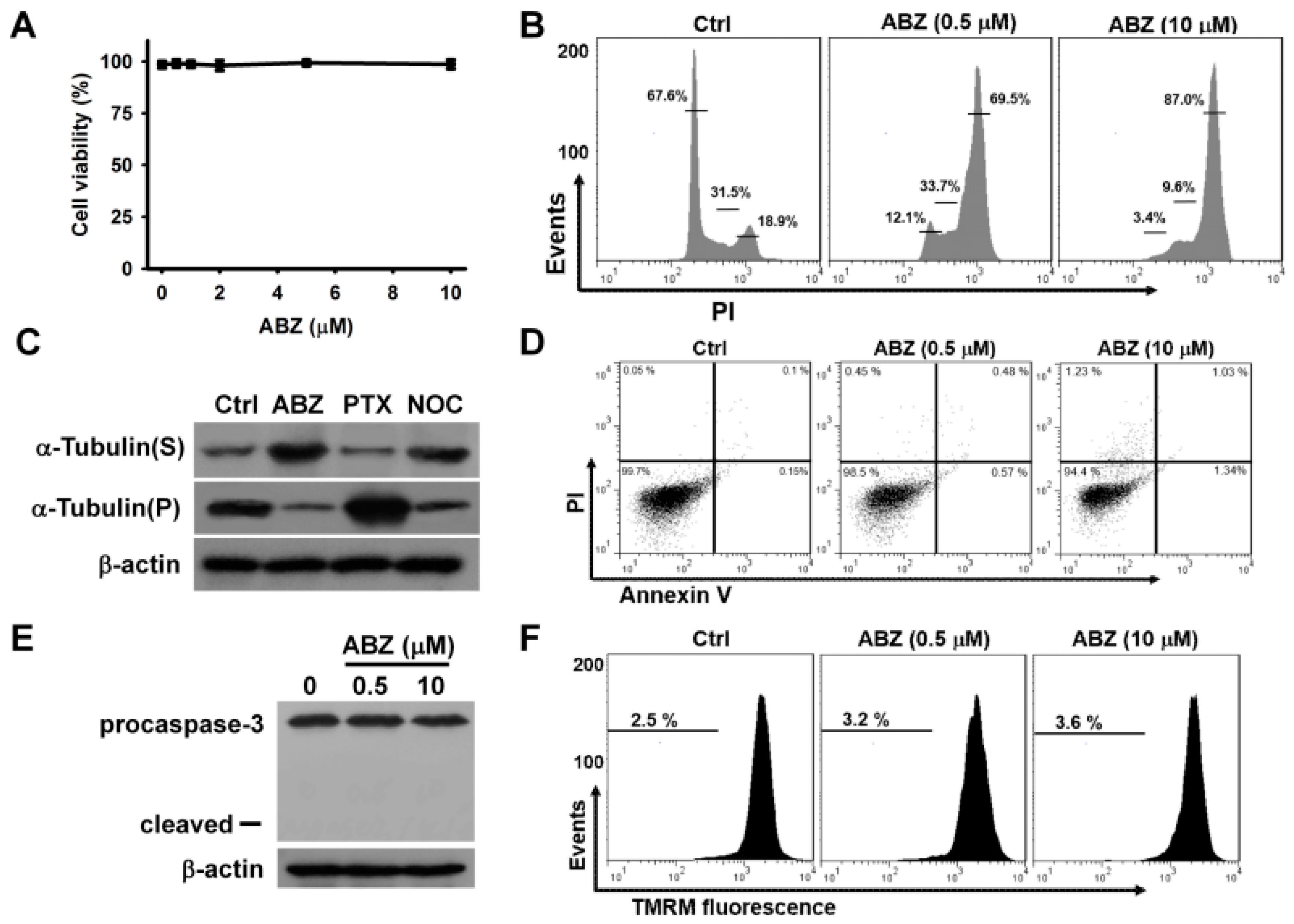
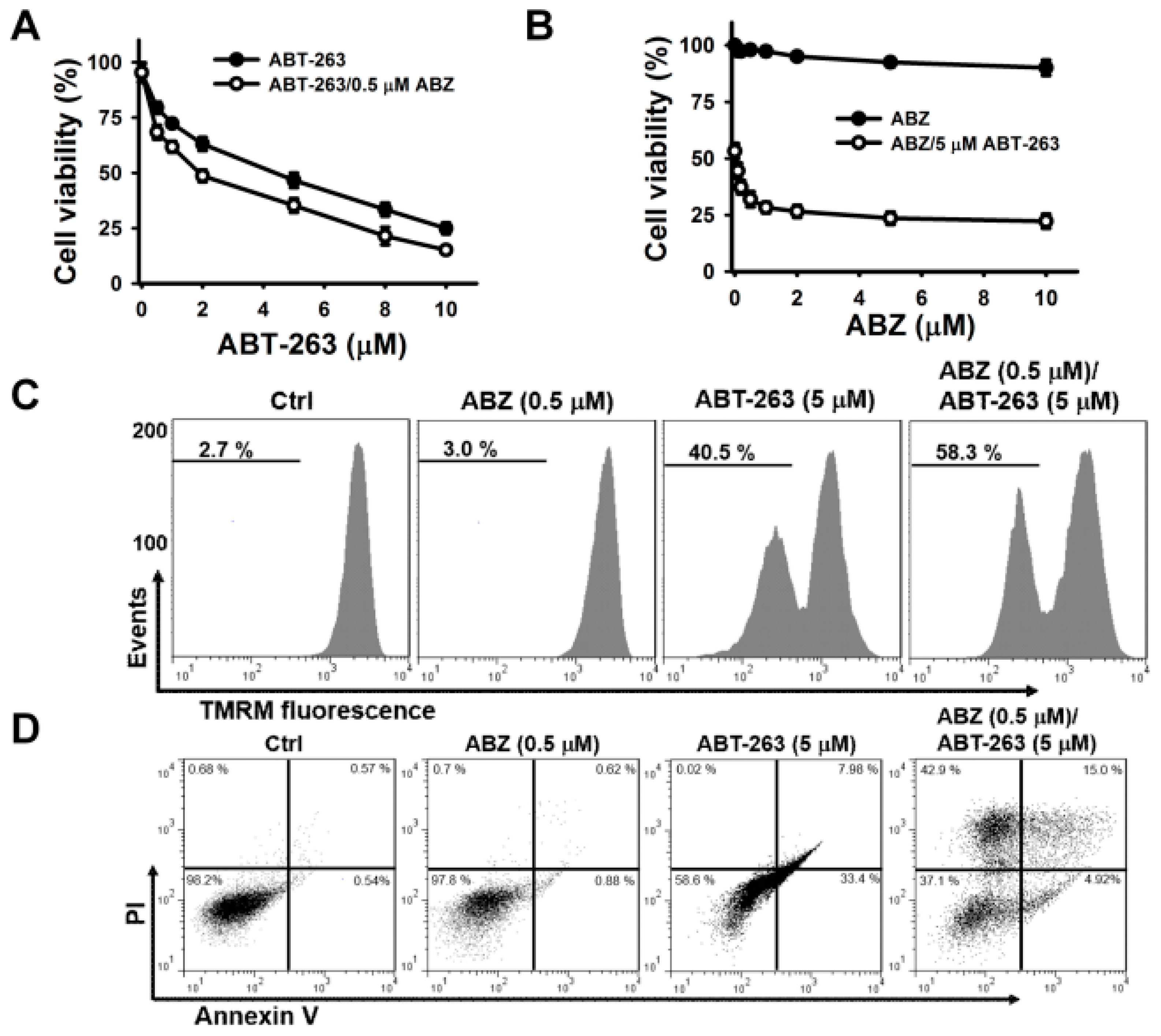
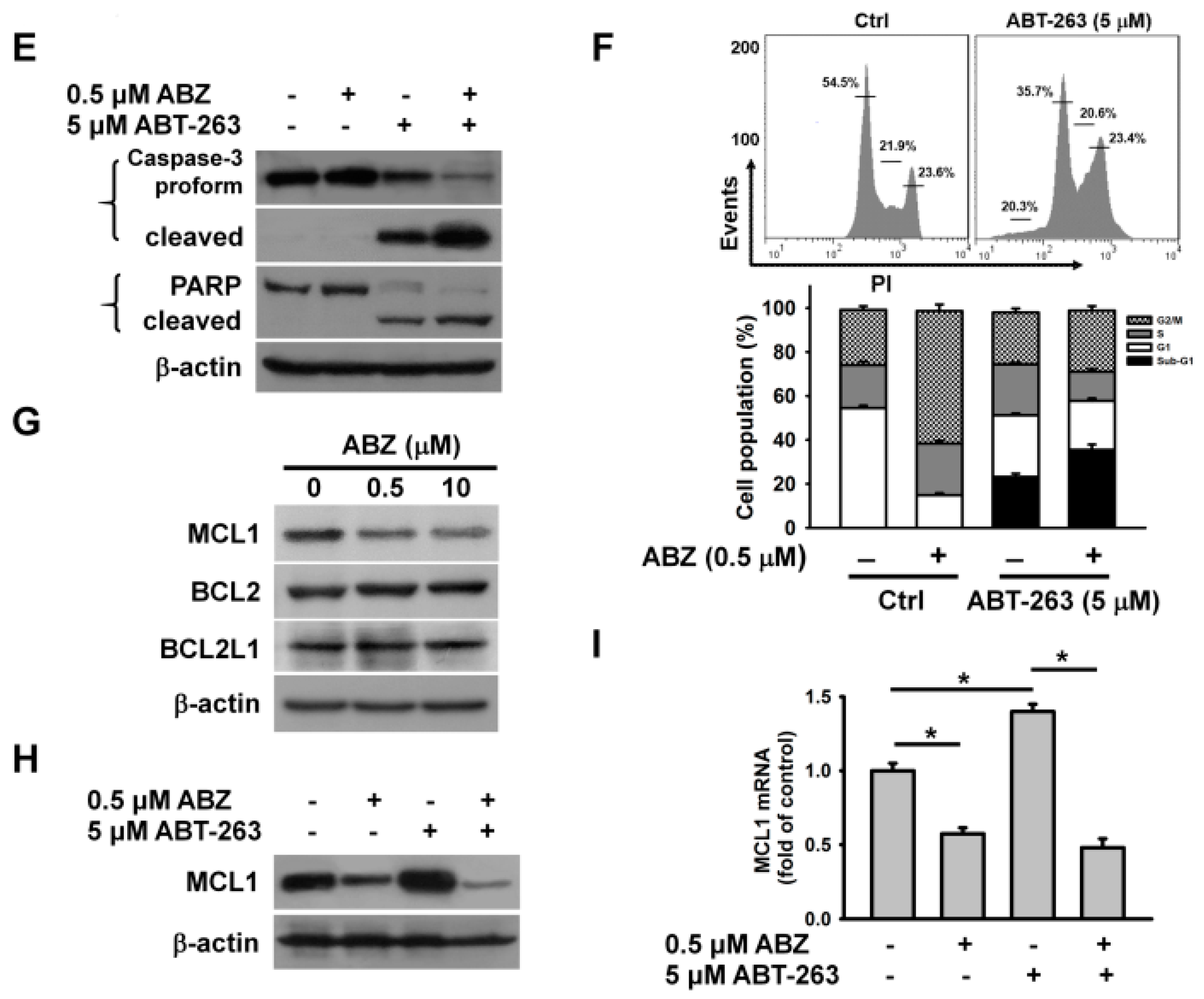
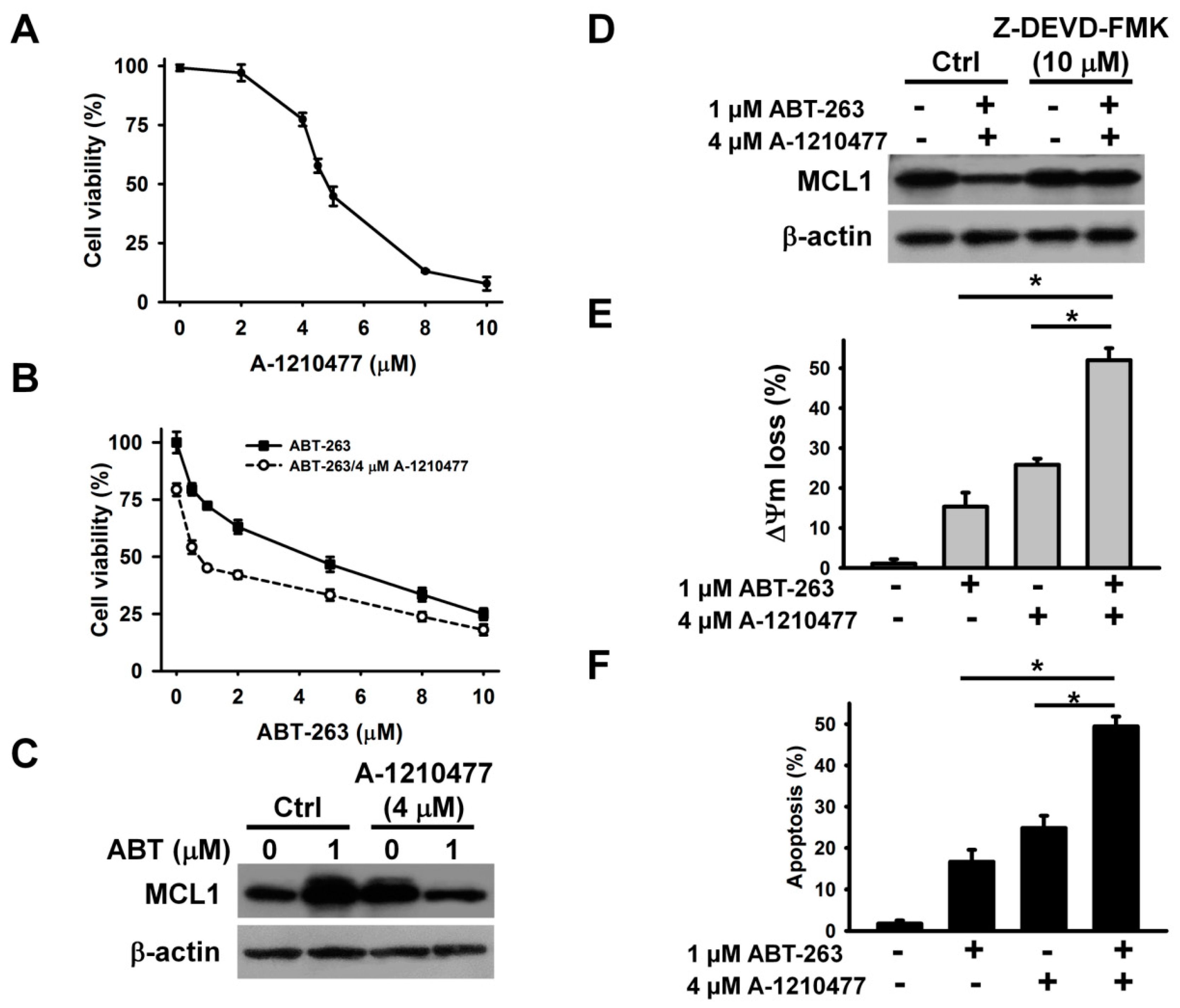
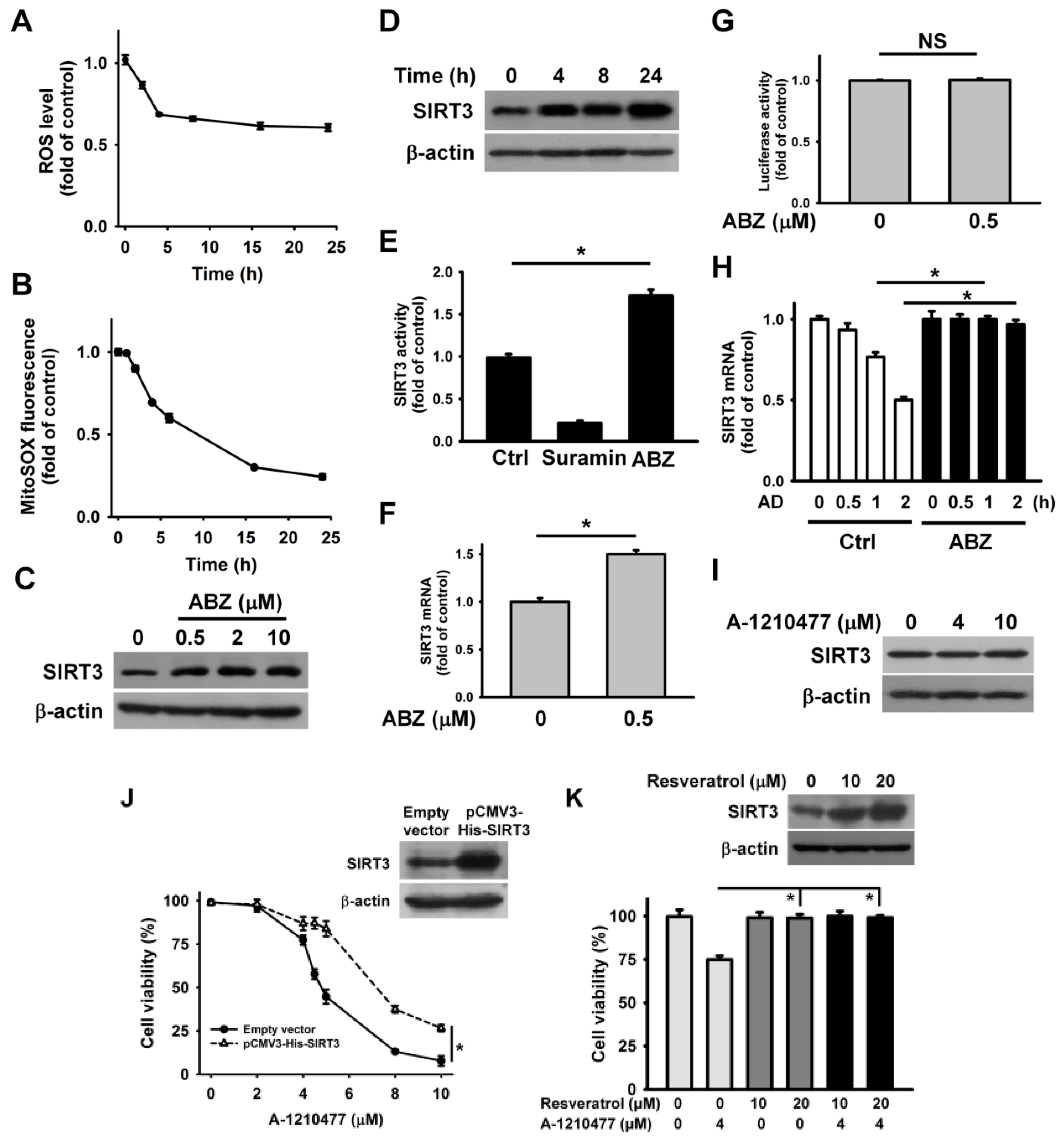
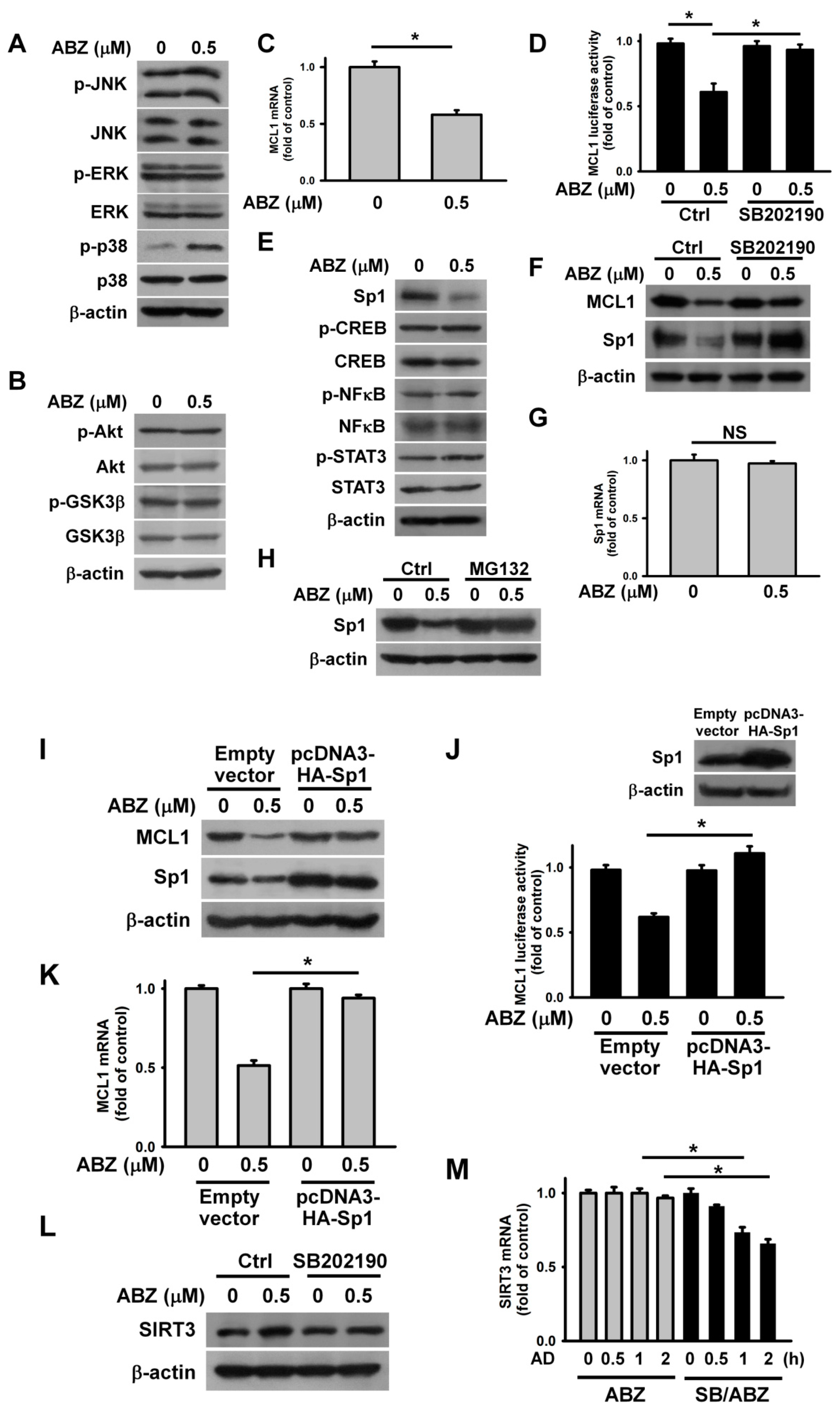
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Cycle Analysis
4.4. Measurement of Intracellular ROS Levels and Mitochondrial Depolarization
4.5. Preparation of Soluble and Insoluble Tubulin Fractions from Cells
4.6. Quantitiative PCR (qPCR)
4.7. Immunoblot Analysis of Protein Expression
4.8. Luciferase Assay
4.9. Meaurement of SIRT3 Deacetylase Activity
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABZ | Albendazole |
| CML | Chronic myeloid leukemia |
| H2DCFDA | Dichlorodihydrofluorescein diacetate |
| FUZ | Flubendazole |
| MBZ | Mebendazole (MBZ) |
| ΔΨm | Mitochondrial membrane potential |
| MTA | Microtubule targeting agents |
| qPCR | Quantitiative PCR |
| TMRM | Tetramethylrhodamine |
References
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, M.O.; Prota, A.E. Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef]
- Köhler, P. The biochemical basis of anthelmintic action and resistance. Int. J. Parasitol. 2001, 31, 336–345. [Google Scholar] [CrossRef]
- Jasmer, D.P.; Yao, C.; Rehman, A.; Johnson, S. Multiple lethal effects induced by a benzimidazole anthelmintic in the anterior intestine of the nematode Haemonchus contortus. Mol. Biochem. Parasitol. 2000, 105, 81–90. [Google Scholar] [CrossRef]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Beheshti Zavareh, R.; et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef] [Green Version]
- Torres, F.C.; García-Rubiño, M.E.; Lozano-López, C.; Kawano, D.F.; Eifler-Lima, V.L.; von Poser, G.L.; Campos, J.M. Imidazoles and benzimidazoles as tubulin-modulators for anti-cancer therapy. Curr. Med. Chem. 2015, 22, 1312–1323. [Google Scholar] [CrossRef]
- Castro, L.S.; Kviecinski, M.R.; Ourique, F.; Parisotto, E.B.; Grinevicius, V.M.; Correia, J.F.; Wilhelm Filho, D.; Pedrosa, R.C. Albendazole as a promising molecule for tumor control. Redox Biol. 2016, 10, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a candidate for drug repurposing in oncology: An extensive review of current literature. Cancers 2019, 11, E1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.; Kim, Y.J.; An, H.; Sung, D.; Cho, T.M.; Farrand, L.; Jang, S.; Seo, J.H.; Kim, J.Y. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int. J. Cancer 2018, 143, 1978–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Zhao, H.; Xie, X.; Luo, M.; Gao, Z.; Sun, H.; Huang, Z. The anthelmintic drug flubendazole induces cell apoptosis and inhibits NF-κB signaling in esophageal squamous cell carcinoma. OncoTargets Ther. 2019, 12, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Bochkur Dratver, M.; Yazal, T.; Dong, K.; Nguyen, A.; Yu, G.; Dao, A.; Bochkur Dratver, M.; Duhachek-Muggy, S.; Bhat, K.; et al. Mebendazole potentiates radiation therapy in triple-negative breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 195–207. [Google Scholar] [CrossRef]
- Wang, L.J.; Lee, Y.C.; Huang, C.H.; Shi, Y.J.; Chen, Y.J.; Pei, S.N.; Chou, Y.W.; Chang, L.S. Non-mitotic effect of albendazole triggers apoptosis of human leukemia cells via SIRT3/ROS/p38 MAPK/TTP axis-mediated TNF-α upregulation. Biochem. Pharmacol. 2019, 162, 154–168. [Google Scholar] [CrossRef]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Haschka, M.D.; Soratroi, C.; Kirschnek, S.; Häcker, G.; Hilbe, R.; Geley, S.; Villunger, A.; Fava, L.L. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat. Commun. 2015, 6, 6891. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, R.M.; Santamaria, B.; Bargellesi, A.; Cosulich, E.; Fabbi, M. Late apoptotic effects of taxanes on K562 erythroleukemia cells: Apoptosis is delayed upstream of caspase-3 activation. Int. J. Cancer 2000, 85, 527–533. [Google Scholar] [CrossRef]
- Aichberger, K.J.; Mayerhofer, M.; Krauth, M.T.; Skvara, H.; Florian, S.; Sonneck, K.; Akgul, C.; Derdak, S.; Pickl, W.F.; Wacheck, V.; et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): Evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005, 105, 3303–3311. [Google Scholar] [CrossRef] [Green Version]
- Lam, L.T.; Zhang, H.; Xue, J.; Leverson, J.D.; Bhathena, A. Antihelminthic benzimidazoles potentiate navitoclax (ABT-263) activity by inducing Noxa-dependent apoptosis in non-small cell lung cancer (NSCLC) cell lines. Cancer Cell Int. 2015, 15, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Ni, Z.; Dai, X.; Qin, L.; Li, X.; Xu, L.; Lian, J.; He, F. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol. Cancer 2014, 13, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedtke, D.A.; Niu, X.; Pan, Y.; Zhao, J.; Liu, S.; Edwards, H.; Chen, K.; Lin, H.; Taub, J.W.; Ge, Y. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct. Target. Ther. 2017, 2, 17012. [Google Scholar] [CrossRef] [PubMed]
- Leverson, J.D.; Zhang, H.; Chen, J.; Tahir, S.K.; Phillips, D.C.; Xue, J.; Nimmer, P.; Jin, S.; Smith, M.; Xiao, Y.; et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015, 6, e1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, Y.; Hall, C.P.; Reynolds, C.P.; Kang, M.H. Caspase-dependent Mcl-1 cleavage and effect of Mcl-1 phosphorylation in ABT-737-induced apoptosis in human acute lymphoblastic leukemia cell lines. Exp. Biol. Med. 2014, 239, 1390–1402. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef]
- Chen, T.; Li, J.; Liu, J.; Li, N.; Wang, S.; Liu, H.; Zeng, M.; Zhang, Y.; Bu, P. Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H424–H434. [Google Scholar] [CrossRef]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Huang, C.H.; Shi, Y.J.; Lee, Y.C.; Wang, L.J.; Chang, L.S. The suppressive effect of arsenic trioxide on TET2-FOXP3-Lyn-Akt axis-modulated MCL1 expression induces apoptosis in human leukemia cells. Toxicol. Appl. Pharmacol. 2018, 358, 43–55. [Google Scholar] [CrossRef]
- Akgul, C.; Turner, P.C.; White, M.R.; Edwards, S.W. Functional analysis of the human MCL-1 gene. Cell. Mol. Life. Sci. 2000, 57, 684–691. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, J.; Yuan, Y.; Xia, Z.; Chen, M.; Xie, L.; Ma, X.; Wang, J.; Ouyang, S.; Wu, Q.; et al. Regulation of Mcl-1 by constitutive activation of NF-κB contributes to cell viability in human esophageal squamous cell carcinoma cells. BMC Cancer 2014, 14, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, D.J.; Soderquist, R.S.; Bates, D.; Eastman, A. Confounding off-target effects of BH3 mimetics at commonly used concentrations: MIM1, UMI-77, and A-1210477. Cell Death Dis. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-mimetic drugs: Blazing the trail for new cancer medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Giralt, A.; Villarroya, F. SIRT3, a pivotal actor in mitochondrial functions: Metabolism, cell death and aging. Biochem. J. 2012, 444, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Liu, B.; Yu, D.; Zuo, Y.; Cai, R.; Yang, J.; Cheng, J. SIRT3 deacetylase activity confers chemoresistance in AML via regulation of mitochondrial oxidative phosphorylation. Br. J. Haematol. 2019, 187, 49–64. [Google Scholar] [CrossRef]
- Liu, W.H.; Chang, L.S. Suppression of Akt/Foxp3-mediated miR-183 expression blocks Sp1-mediated ADAM17 expression and TNFα-mediated NFκB activation in piceatannol-treated human leukemia U937 cells. Biochem. Pharmacol. 2012, 84, 670–680. [Google Scholar] [CrossRef]
- Lee, Y.C.; Wang, L.J.; Huang, C.H.; Shi, Y.J.; Chang, L.S. ABT-263-induced MCL1 upregulation depends on autophagy-mediated 4EBP1 downregulation in human leukemia cells. Cancer Lett. 2018, 432, 191–204. [Google Scholar] [CrossRef]
- Wang, L.J.; Chiou, J.T.; Lee, Y.C.; Huang, C.H.; Shi, Y.J.; Chang, L.S. SIRT3, PP2A and TTP protein stability in the presence of TNF-α on vincristine-induced apoptosis of leukaemia cells. J. Cell. Mol. Med. 2020, 24, 2552–2565. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.-J.; Liou, L.-R.; Shi, Y.-J.; Chiou, J.-T.; Lee, Y.-C.; Huang, C.-H.; Huang, P.-W.; Chang, L.-S. Albendazole-Induced SIRT3 Upregulation Protects Human Leukemia K562 Cells from the Cytotoxicity of MCL1 Suppression. Int. J. Mol. Sci. 2020, 21, 3907. https://doi.org/10.3390/ijms21113907
Wang L-J, Liou L-R, Shi Y-J, Chiou J-T, Lee Y-C, Huang C-H, Huang P-W, Chang L-S. Albendazole-Induced SIRT3 Upregulation Protects Human Leukemia K562 Cells from the Cytotoxicity of MCL1 Suppression. International Journal of Molecular Sciences. 2020; 21(11):3907. https://doi.org/10.3390/ijms21113907
Chicago/Turabian StyleWang, Liang-Jun, Li-Ren Liou, Yi-Jun Shi, Jing-Ting Chiou, Yuan-Chin Lee, Chia-Hui Huang, Po-Wei Huang, and Long-Sen Chang. 2020. "Albendazole-Induced SIRT3 Upregulation Protects Human Leukemia K562 Cells from the Cytotoxicity of MCL1 Suppression" International Journal of Molecular Sciences 21, no. 11: 3907. https://doi.org/10.3390/ijms21113907
APA StyleWang, L. -J., Liou, L. -R., Shi, Y. -J., Chiou, J. -T., Lee, Y. -C., Huang, C. -H., Huang, P. -W., & Chang, L. -S. (2020). Albendazole-Induced SIRT3 Upregulation Protects Human Leukemia K562 Cells from the Cytotoxicity of MCL1 Suppression. International Journal of Molecular Sciences, 21(11), 3907. https://doi.org/10.3390/ijms21113907