Targeted Delivery of Mesenchymal Stem Cell-Derived Nanovesicles for Spinal Cord Injury Treatment

 , ,
, ,  and
and 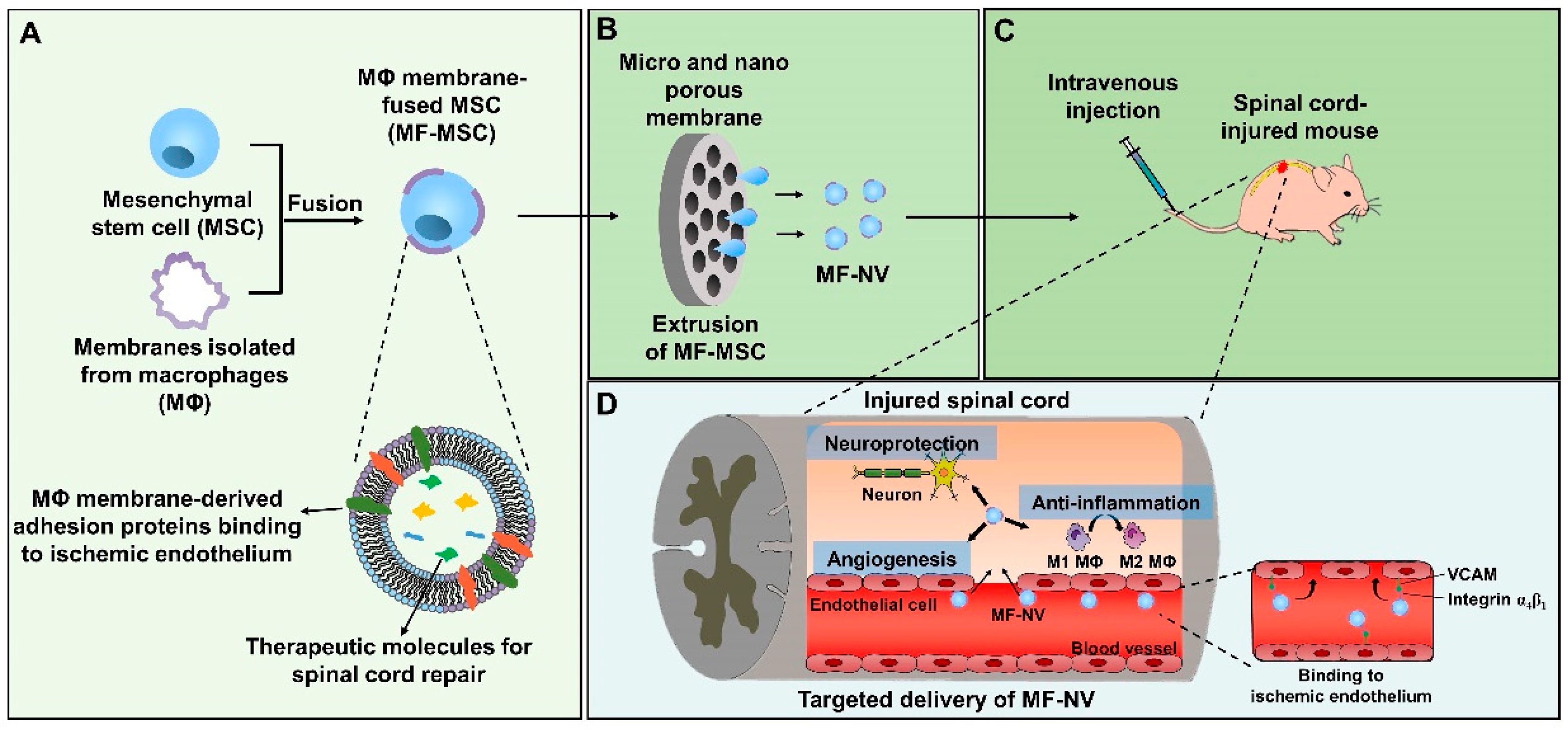{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
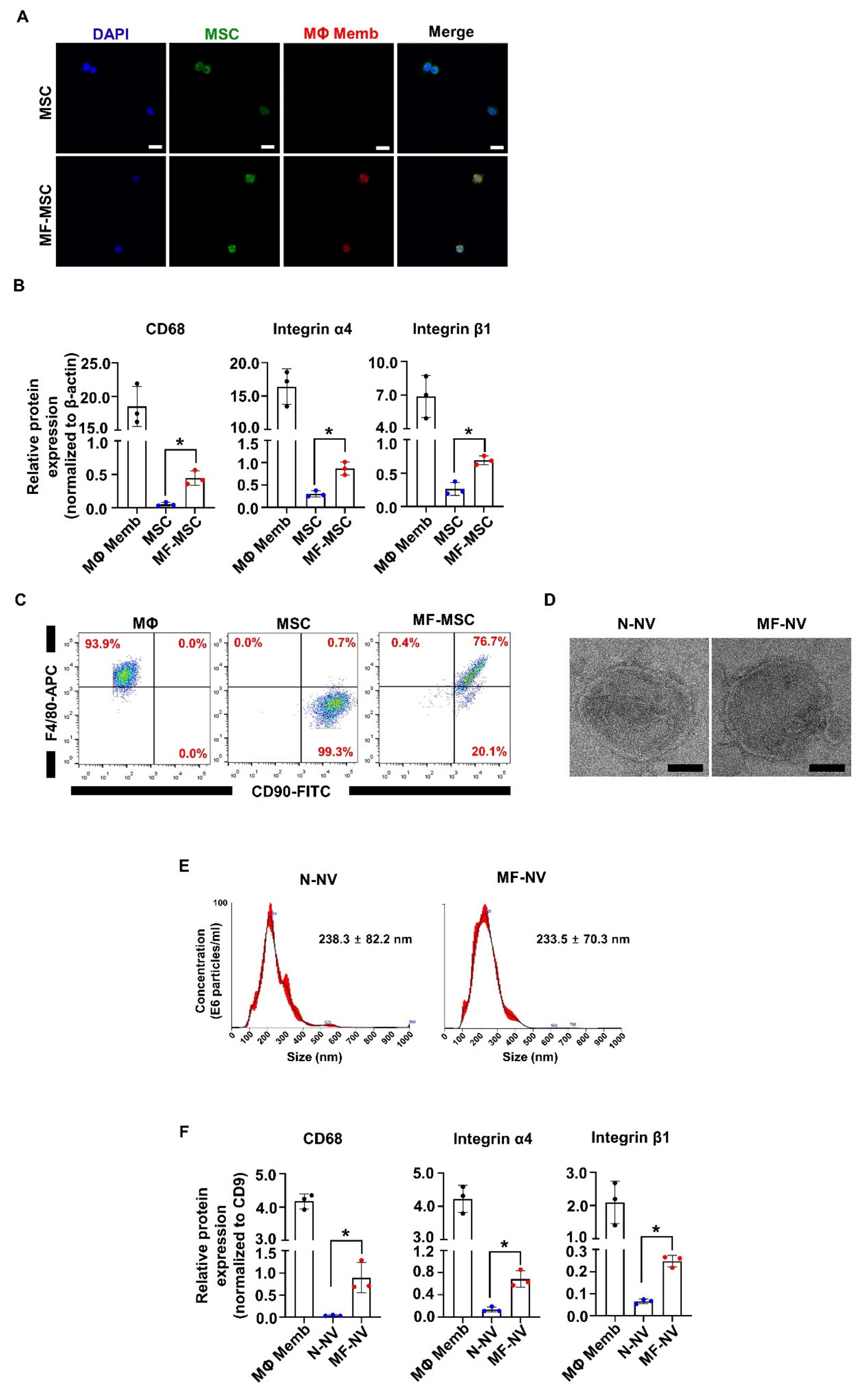
2.1. Characterization of MF-MSCs and MF-NVs
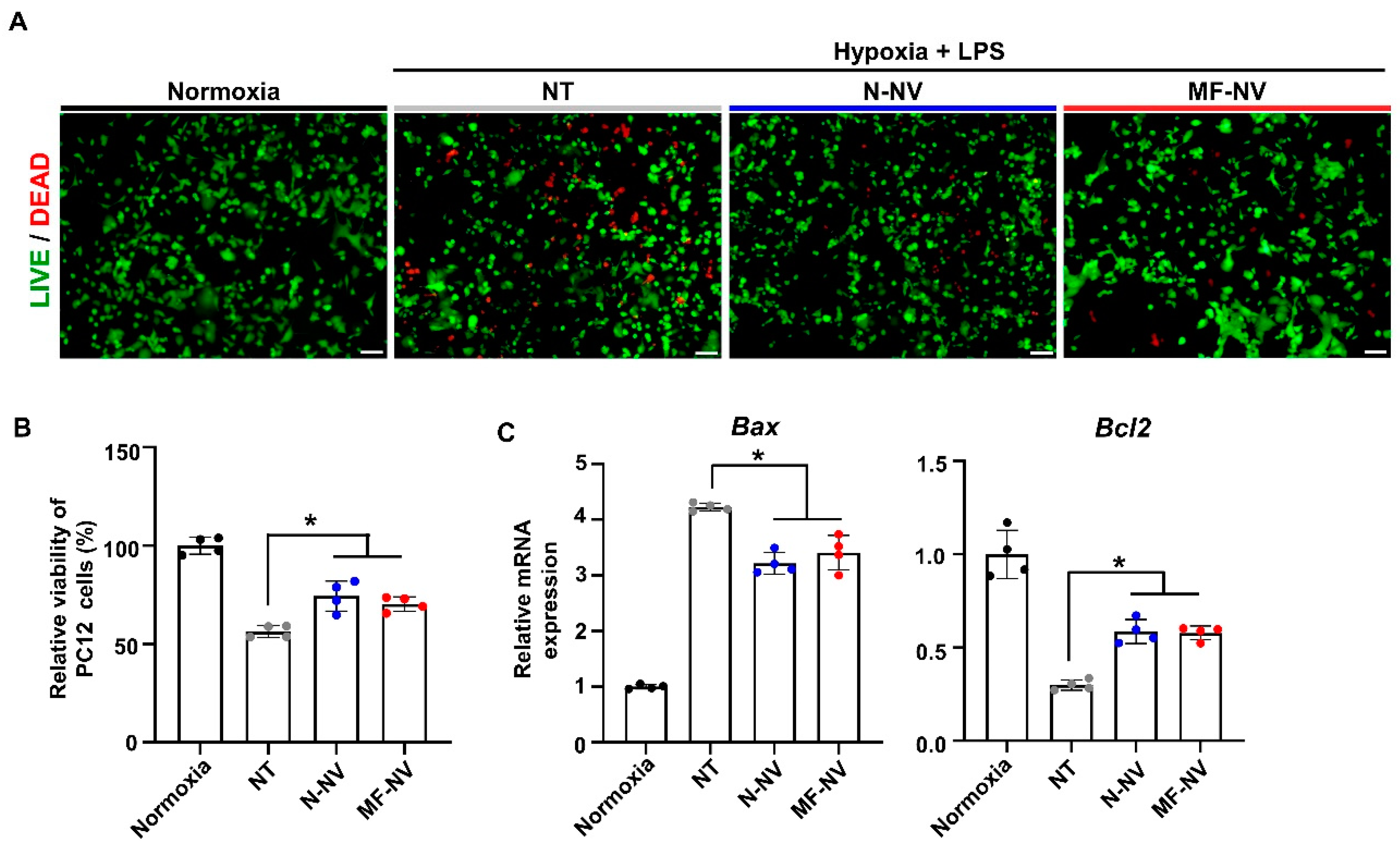
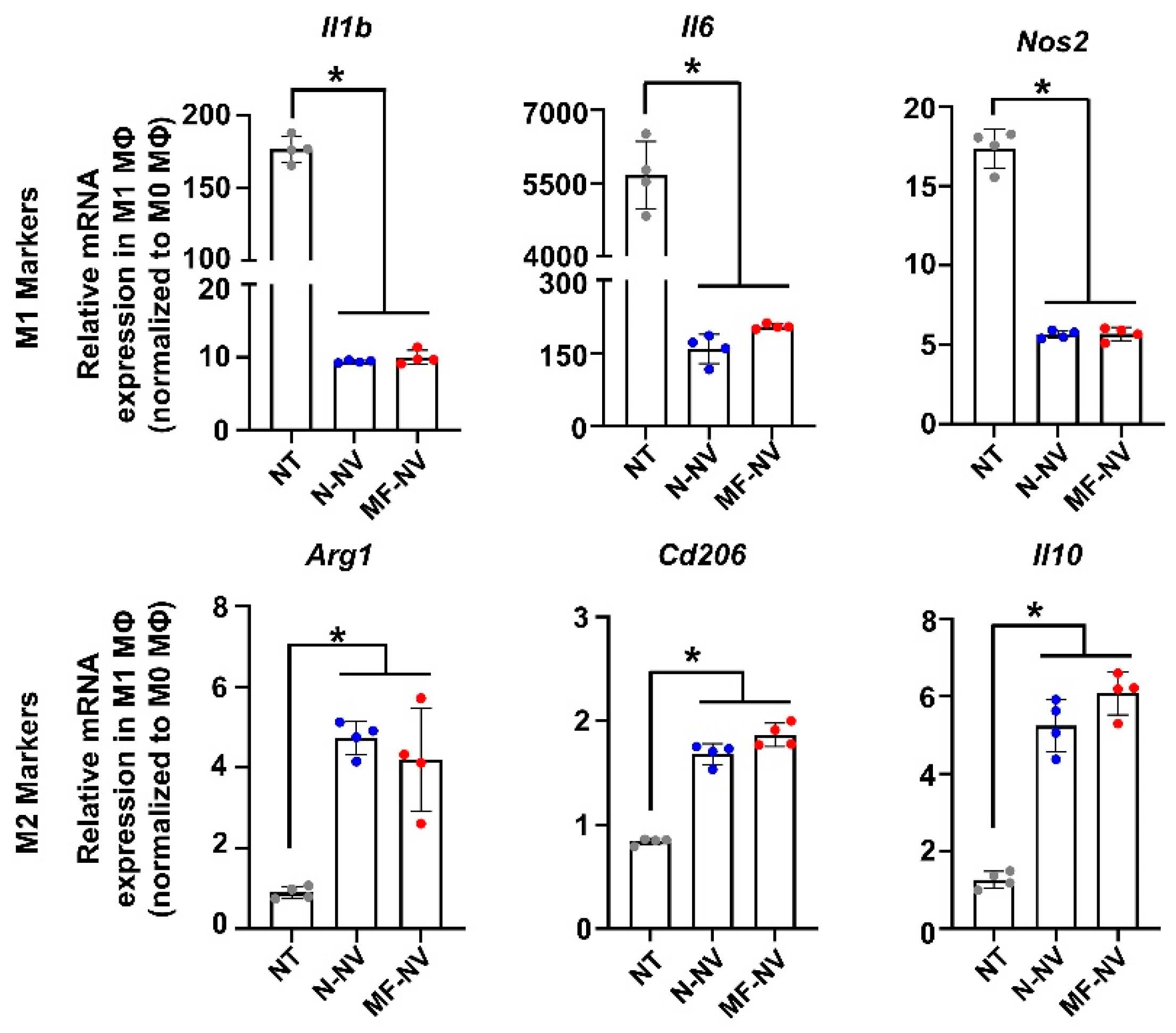
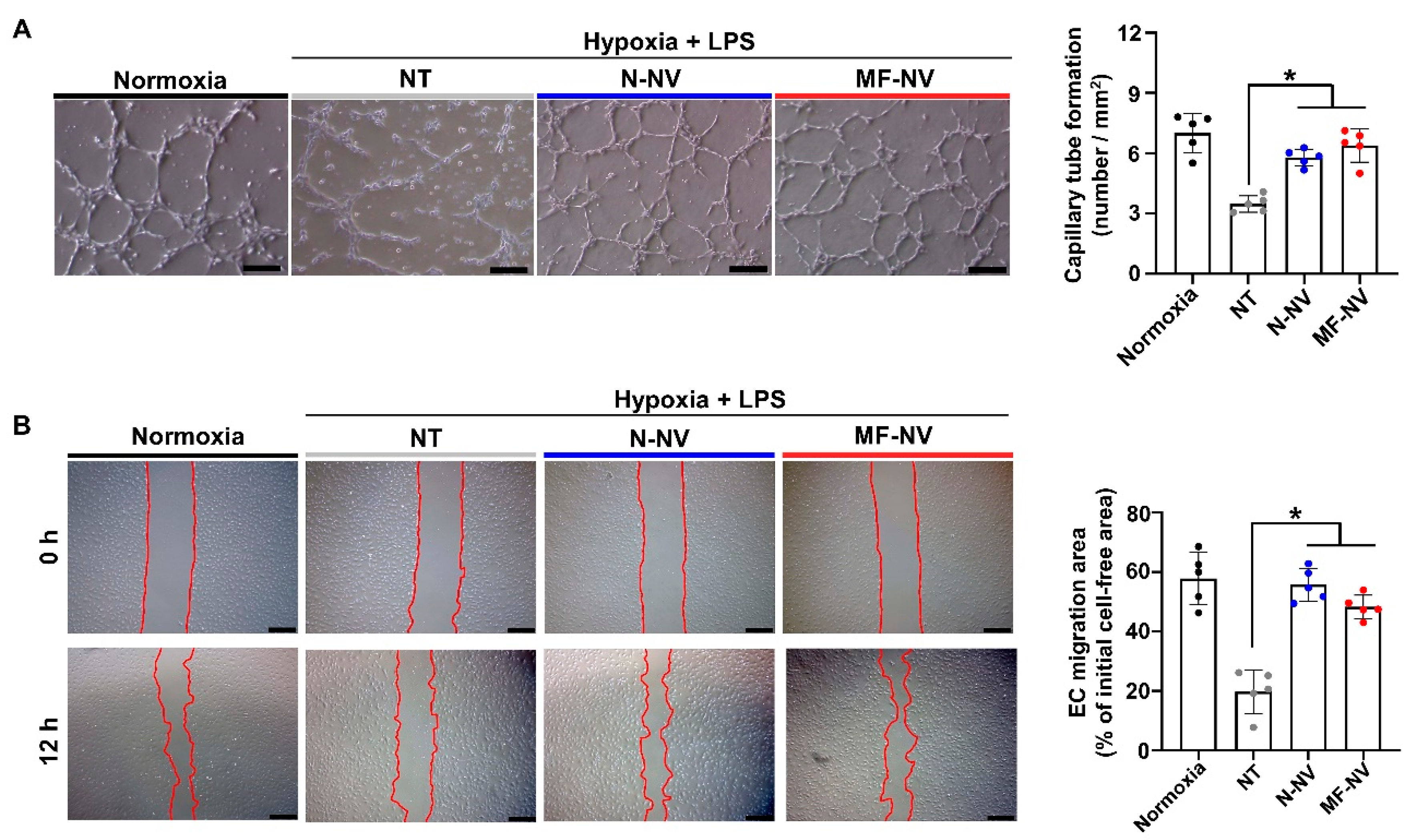
2.2. Anti-Apoptotic, Anti-Inflammatory, and Angiogenic Effect of MF-NV In Vitro
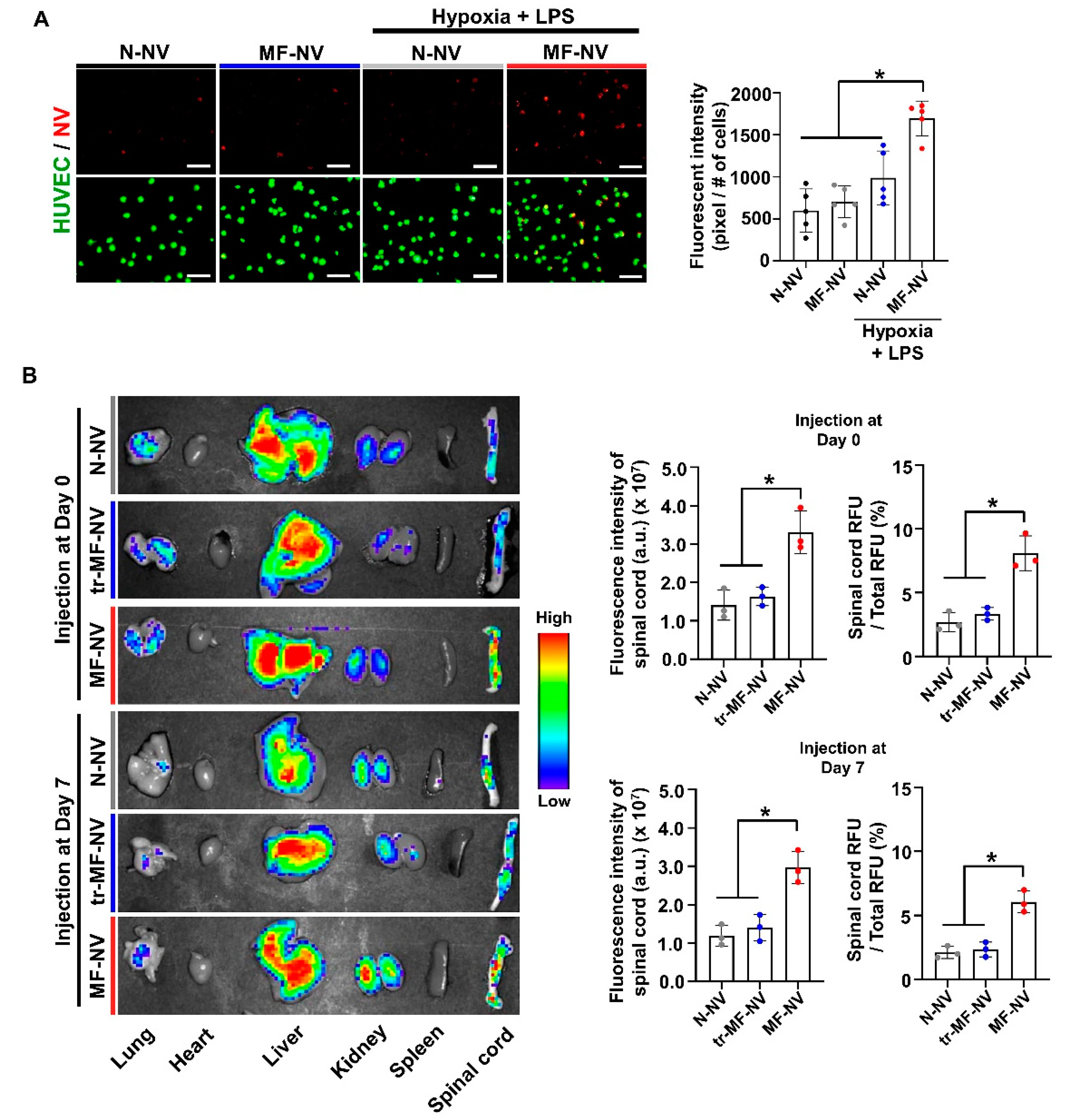
2.3. Enhanced Targeting Efficiency of MF-NVs In Vitro and In Vivo
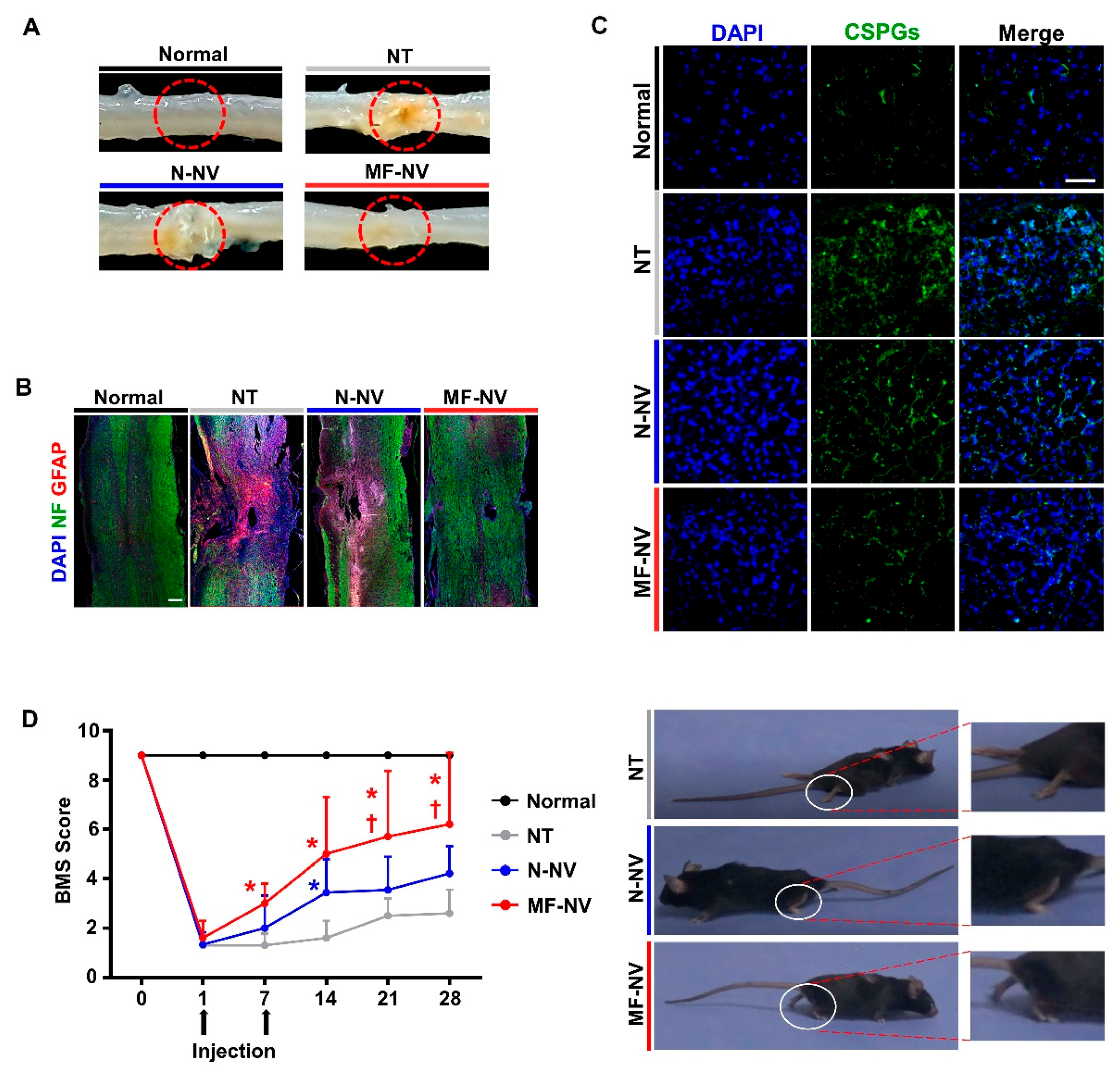
2.4. Reduced Glial Scar Formation and Improved Function Recovery by MF-NVs In Vivo
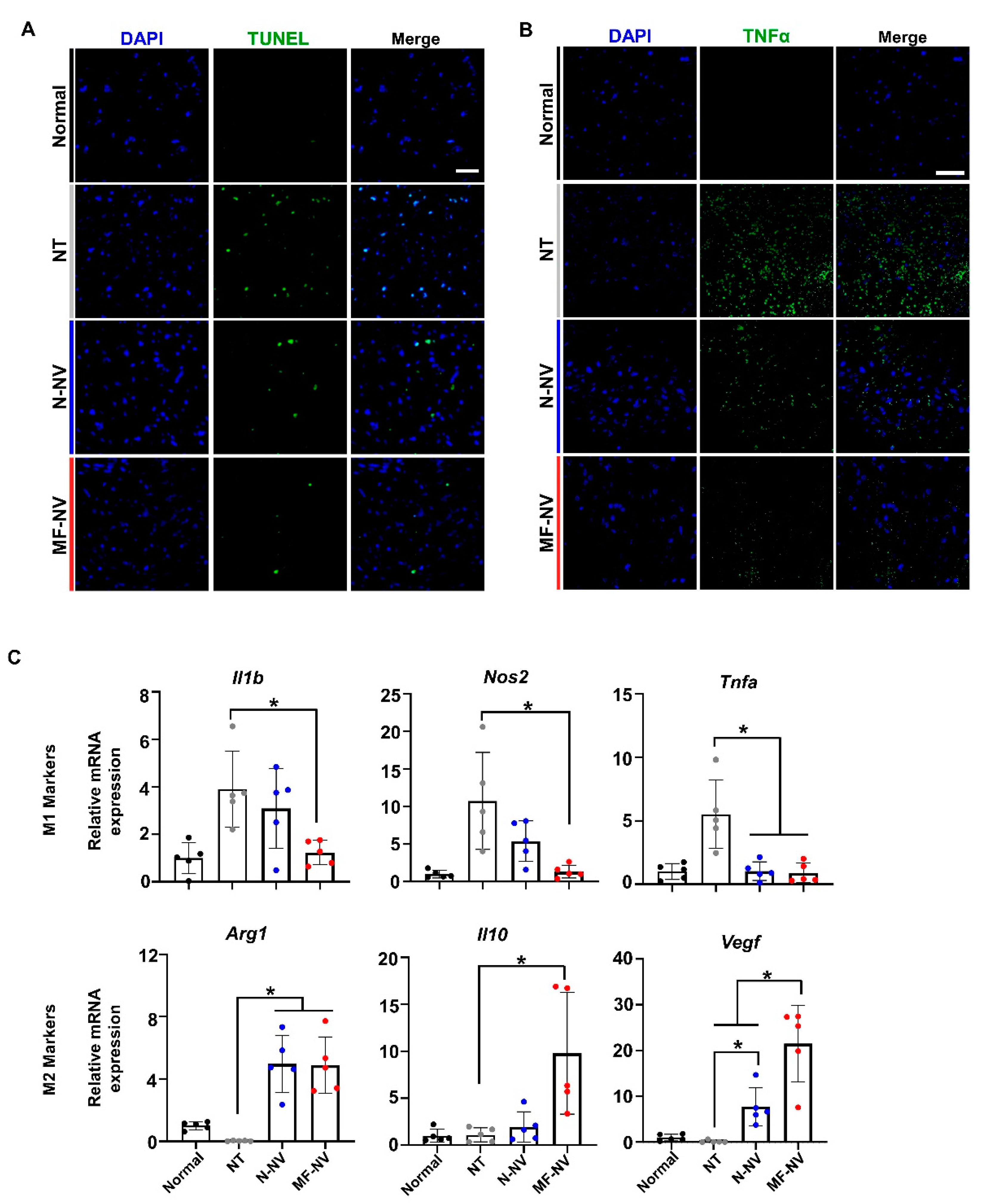
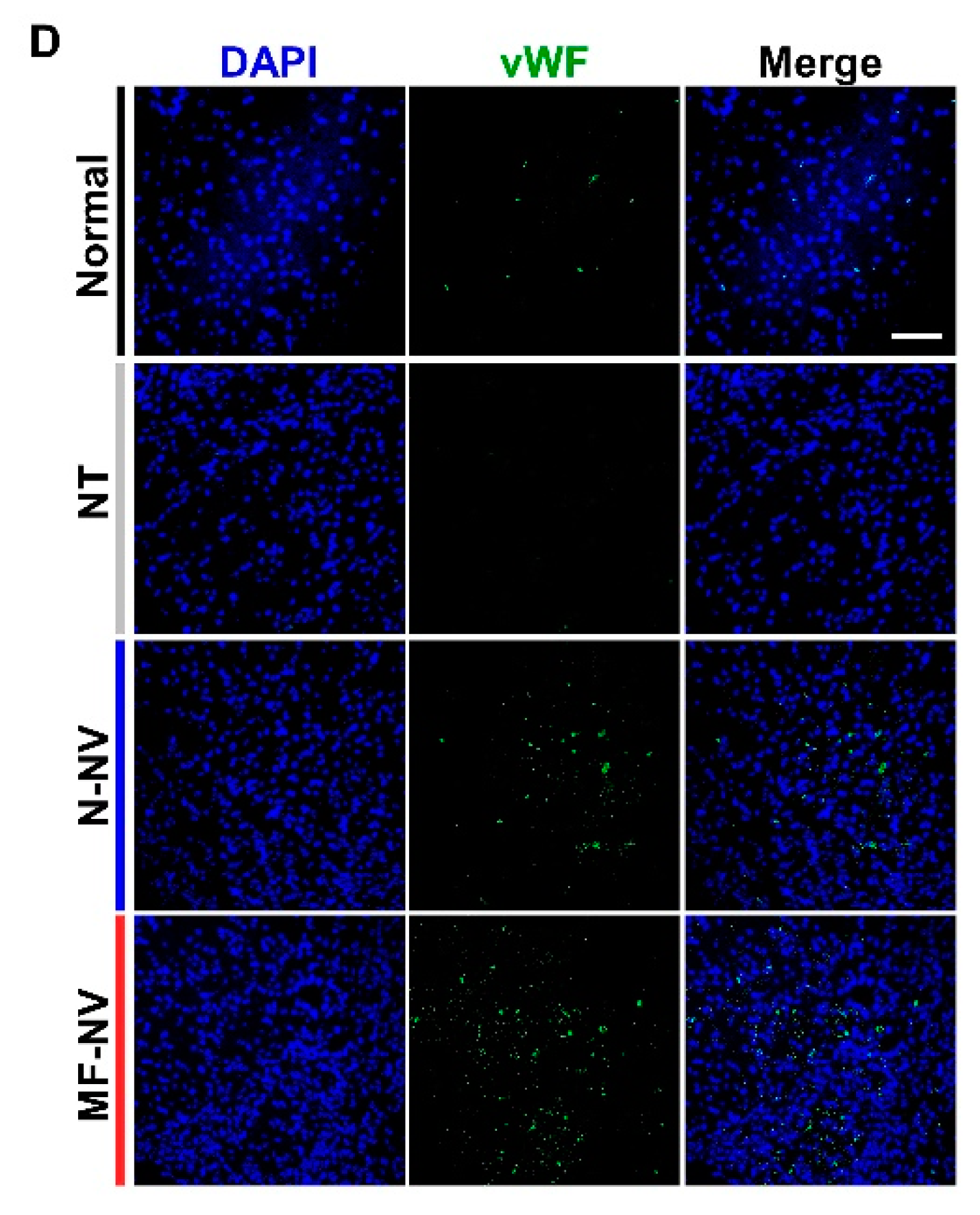
2.5. Enhanced Neuroprotection, Anti-Inflammation, and Angiogenesis by MF-NVs In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Isolation of Macrophage Membranes
4.3. Generation and Characterization of MF-MSCs
4.4. Generation and Characterization of MF-NVs
4.5. Western Blot Analysis
4.6. Flow Cytometry
4.7. In Vitro Therapeutic Effects of MF-NVs
4.8. Quantitative PCR
4.9. In Vitro NV Binding Assay
4.10. SCI Model and Treatment
4.11. Ex Vivo Biodistribution of NVs
4.12. Immunohistochemical Assessment In Vivo
4.13. In Vivo Apoptosis Assessment
4.14. Behavior Evaluation
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SCI | Spinal cord injury |
| MSC | Mesenchymal stem cell |
| NV | Exosome-mimetic nanovesicle |
| VLA4 | Very late antigen 4 |
| FBS | Fetal bovine serum |
| PBS | Phosphate buffered saline |
| LPS | Lipopolysaccharide |
| ERK | Extracellular signal-regulated kinases |
| AKT | Serine/threonine-specific protein kinase |
| PI3K | Phosphatidylinositol 3-kinases |
| PCNA | Proliferating cell nuclear antigen |
| HUVEC | Human umbilical vein endothelial cell |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| vWF | von Willebrand factor |
| Arg1 | Arginase 1 |
| NF | Neurofilament |
| GFAP | Glial fibrillary acidic protein |
| CSPG | chondroitin sulfate proteoglycans |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| TNFα | Tumor necrosis factor alpha |
| BMS | Basso mouse scale |
| PEG | Polyethylene glycol |
| TEM | Transmission electron microscopy |
| FACS | Fluorescence-activated cell sorting |
| IL | Interleukin |
| Nos2 | Nitric oxide synthase 2 |
| IHC | Immunohistochemistry |
| Vegf | Vascular endothelial growth factor |
| ECM | Extracellular matrix |
References
- McDonald, J.W.; Sadowsky, C. Spinal-cord injury. The Lancet 2002, 359, 417–425. [Google Scholar] [CrossRef]
- De Almeida, F.M.; Marques, S.A.; Ramalho, B.D.; Massoto, T.B.; Martinez, A.M.B. Chronic spinal cord lesions respond positively to tranplants of mesenchymal stem cells. Restor. Neurol. Neurosci. 2015, 33, 43–55. [Google Scholar] [CrossRef]
- Sasaki, M.; Radtke, C.; Tan, A.M.; Zhao, P.; Hamada, H.; Houkin, K.; Honmou, O.; Kocsis, J.D. BDNF-hypersecreting human mesenchymal stem cells promote functional recovery, axonal sprouting, and protection of corticospinal neurons after spinal cord Injury. J. Neurosci. 2009, 29, 14932–14941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spejo, A.B.; Carvalho, J.L.; Goes, A.M.; Oliveira, A.L.R. Neuroprotective effects of mesenchymal stem cells on spinal motoneurons following ventral root axotomy: Synapse stability and axonal regeneration. Neuroscience 2013, 250, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Uchida, K.; Guerrero, A.R.; Watanabe, S.; Sugita, D.; Takeura, N.; Yoshida, A.; Long, G.; Wright, K.T.; Johnson, W.E.B.; et al. Transplantation of mesenchymal stem cells promotes an alternative pathway of macrophage activation and functional recovery after spinal cord injury. J. Neurotrauma 2012, 29, 1614–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quertainmont, R.; Cantinieaux, D.; Botman, O.; Sid, S.; Schoenen, J.; Franzen, R. Mesenchymal stem cell graft improves recovery after spinal cord injury in adult rats through neurotrophic and pro-angiogenic actions. PLoS ONE 2012, 7, e39500. [Google Scholar] [CrossRef] [PubMed]
- Bhang, S.H.; Lee, Y.E.; Cho, S.W.; Shim, J.W.; Lee, S.H.; Choi, C.Y.; Chang, J.W.; Kim, B.S. Basic fibroblast growth factor promotes bone marrow stromal cell transplantation-mediated neural regeneration in traumatic brain injury. Biochem. Biophys. Res. Commun. 2007, 359, 40–45. [Google Scholar] [CrossRef]
- Mun, C.H.; Kang, M.I.; Shin, Y.D.; Kim, Y.; Park, Y.B. The expression of immunomodulation-related cytokines and genes of adipose- and bone marrow-derived human mesenchymal stromal cells from early to late passages. Tissue Eng. Regen. Med. 2018, 15, 771–779. [Google Scholar] [CrossRef]
- Kim, J.W.; Ha, K.Y.; Molon, J.N.; Kim, Y.H. Bone marrow-derived mesenchymal stem cell transplantation for chronic spinal cord injury in rats comparative study between intralesional and intravenous transplantation. Spine 2013, 38, E1065–E1074. [Google Scholar] [CrossRef]
- Boido, M.; Garbossa, D.; Fontanella, M.; Ducati, A.; Vercelli, A. Mesenchymal stem cell transplantation reduces glial cyst and improves functional outcome after spinal cord compression. World Neurosurg. 2014, 81, 183–190. [Google Scholar] [CrossRef]
- Willerth, S.M.; Sakiyama-Elbert, S.E. Cell therapy for spinal cord regeneration. Adv. Drug Deliv. Rev. 2008, 60, 263–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casiraghi, F.; Remuzzi, G.; Abbate, M.; Perico, N. Multipotent mesenchymal stromal cell therapy and risk of malignancies. Stem Cell Rev. Rep. 2013, 9, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Chudickova, M.; Vackova, I.; Urdzikova, L.M.; Jancova, P.; Kekulova, K.; Rehorova, M.; Turnovcova, K.; Jendelova, P.; Kubinova, S. The effect of wharton jelly-derived mesenchymal stromal cells and their conditioned media in the treatment of a rat spinal cord injury. Int. J. Mol. Sci. 2019, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Huang, J.; Geng, Y.J.; Qian, H.Y.; Wang, F.; Liu, X.H.; Shang, M.S.; Nie, S.P.; Liu, N.A.; Du, X.; et al. Paracrine action of mesenchymal stem cells revealed by single cell gene profiling in infarcted murine hearts. PLoS ONE 2015, 10, e0129164. [Google Scholar] [CrossRef] [Green Version]
- Vizoso, F.J.; Eiro, N.; Cid, S.; Schneider, J.; Perez-Fernandez, R. Mesenchymal stem cell secretome: Toward cell-free therapeutic strategies in regenerative medicine. Int. J. Mol. Sci. 2017, 18, 1852. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.; Kim, H.S. Exosomes as therapeutic vehicles for cancer. Tissue Eng. Regen. Med. 2019, 16, 213–223. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadaran, P.; Rajendran, R.L.; Lee, H.W.; Kalimuthu, S.; Hong, C.M.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Extracellular vesicles from mesenchymal stem cells activates VEGF receptors and accelerates recovery of hindlimb ischemia. J. Control. Release 2017, 264, 112–126. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.D.; Li, G.Q.; Li, D.H.; Huang, W.J.; Zhang, R.W.; Zhang, H.; Duan, Y.Y.; Wang, B.C. hucMSC derived exosomes promote functional recovery in spinal cord injury mice via attenuating inflammation. Mater. Sci. Eng. C 2018, 89, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.M.; Chen, L.; Chen, W.Q.; Yang, J.J.; Yang, Z.Y.; Shen, Z.Y. Mesenchymal stem cell-derived exosomes improve the microenvironment of infarcted myocardium contributing to angiogenesis and anti-inflammation. Cell. Physiol. Biochem. 2015, 37, 2415–2424. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Zhao, X.N.; Chen, X.N.; Wei, Y.Z.; Du, W.; Wang, Y.B.; Liu, L.A.; Zhao, W.A.; Han, Z.B.; Kong, D.L.; et al. Enhanced therapeutic effects of mesenchymal stem cell-derived exosomes with an unjectable hydrogel for hindlimb uschemia treatment. ACS Appl. Mater. Interfaces 2018, 10, 30081–30091. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.Q.; Li, Y.; Cui, Y.S.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J. Cereb. Blood Flow Metab. 2013, 33, 1711–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.H.; Yin, X.M.; Xu, Y.; Xu, C.C.; Lin, X.; Ye, F.B.; Cao, Y.; Lin, F.Y. Systemic Administration of Exosomes released from mesenchymal stromal cells attenuates apoptosis, inflammation, and promotes angiogenesis after spinal cord injury in rats. J. Neurotrauma 2017, 34, 3388–3396. [Google Scholar] [CrossRef]
- Zhang, B.; Yeo, R.W.Y.; Lai, R.C.; Sim, E.W.K.; Chin, K.C.; Lim, S.K. Mesenchymal stromal cell exosome-enhanced regulatory T-cell production through an antigen-presenting cell-mediated pathway. Cytotherapy 2018, 20, 687–696. [Google Scholar] [CrossRef]
- Ziegler, M.; Haigh, K.; Nguyen, T.; Wang, X.W.; Lim, B.; Yap, M.L.; Eddy, E.M.; Haigh, J.J.; Peter, K. The pulmonary microvasculature entraps induced vascular progenitor cells (iVPCs) systemically delivered after cardiac ischemia-reperfusion injury: Indication for preservation of heart function via paracrine effects beyond engraftment. Microcirculation 2019, 26, e12493. [Google Scholar] [CrossRef]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]
- Jo, W.; Jeong, D.; Kim, J.; Park, J. Self-Renewal of Bone Marrow Stem Cells by Nanovesicles Engineered from Embryonic Stem Cells. Adv. Healthcare Mater. 2016, 5, 3148–3156. [Google Scholar] [CrossRef]
- Oh, K.; Kim, S.R.; Kim, D.K.; Seo, M.W.; Lee, C.; Lee, H.M.; Oh, J.E.; Choi, E.Y.; Lee, D.S.; Gho, Y.S.; et al. In vivo differentiation of therapeutic insulin-producing cells from bone marrow cells via extracellular vesicle-mimetic nanovesicles. ACS Nano 2015, 9, 11718–11727. [Google Scholar] [CrossRef]
- Jo, W.; Kim, J.; Yoon, J.; Jeong, D.; Cho, S.; Jeong, H.; Yoon, Y.J.; Kim, S.C.; Gho, Y.S.; Park, J. Large-scale generation of cell-derived nanovesicles. Nanoscale 2014, 6, 12056–12064. [Google Scholar] [CrossRef] [Green Version]
- Wiklander, O.P.B.; Nordin, J.Z.; O′Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.A.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, R.; Feigelson, S. From rolling to arrest on blood vessels: Leukocyte tap dancing on endothelial integrin ligands and chemokines at sub-second contacts. Semin. Immunol. 2002, 14, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Z.; Dehaini, D.; Zhang, Y.; Zhou, J.L.; Chen, X.Y.; Zhang, L.F.; Fang, R.H.; Gao, W.W.; Zhang, L.F. Neutrophil membrane-coated nanoparticles inhibit synovial inflammation and alleviate joint damage in inflammatory arthritis. Nat. Nanotechnol. 2018, 13, 1182. [Google Scholar] [CrossRef]
- Thamphiwatana, S.; Angsantikul, P.; Escajadillo, T.; Zhang, Q.Z.; Olson, J.; Luk, B.T.; Zhang, S.; Fang, R.H.; Gao, W.W.; Nizet, V.; et al. Macrophage-like nanoparticles concurrently absorbing endotoxins and proinflammatory cytokines for sepsis management. Proc. Natl. Acad. Sci. USA 2017, 114, 11488–11493. [Google Scholar] [CrossRef] [Green Version]
- Beekhuizen, H.; Vanfurth, R. Monocyte adherence to human vascular endothelium. J. Leukoc. Biol. 1993, 54, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.W.; Kang, M.; Kim, H.Y.; Han, J.; Kang, S.; Lee, J.R.; Jeong, G.J.; Kwon, S.P.; Song, S.Y.; Go, S.; et al. M1 macrophage-derived nanovesicles potentiate the anticancer efficacy of immune checkpoint inhibitors. ACS Nano 2018, 12, 8977–8993. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kumar, H.; Jo, M.J.; Kim, J.; Yoon, J.K.; Lee, J.R.; Kang, M.; Choo, Y.W.; Song, S.Y.; Kwon, S.P.; et al. Therapeutic efficacy-potentiated and diseased organ-targeting nanovesicles derived from mesenchymal stem cells for spinal cord injury treatment. Nano Lett. 2018, 18, 4965–4975. [Google Scholar] [CrossRef]
- Li, S.Y.; Qi, Y.; Hu, S.H.; Piao, F.Y.; Guan, H.; Wang, Z.M.; Chen, R.L.; Liu, S. Mesenchymal stem cells-conditioned medium protects PC12 cells against 2,5-hexanedione-induced apoptosis via inhibiting mitochondria-dependent caspase 3 pathway. Toxicol. Ind. Health 2017, 33, 107–118. [Google Scholar] [CrossRef]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal stem cell-derived extracellular vesicles as mediators of anti-inflammatory effects: Endorsement of macrophage polarization. Stem Cell Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.Y.; Lie, P.C.; Miao, T.Y.; Yu, M.X.; Lu, Q.; Feng, T.; Li, J.R.; Zu, T.T.; Liu, X.H.; Li, H. Conditioned medium from umbilical cord mesenchymal stem cells induces migration and angiogenesis. Mol. Med. Rep. 2015, 12, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.S.; Chua, C.C.; Gao, F.P.; Chua, K.W.; Wang, H.; Hamdy, R.C.; Chua, B.H.L. Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain Res. 2008, 1227, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.R.; Park, B.W.; Kim, J.; Choo, Y.W.; Kim, H.Y.; Yoon, J.K.; Kim, H.; Hwang, J.W.; Kang, M.; Kwon, S.P.; et al. Nanovesicles derived from iron oxide nanoparticles-incorporated mesenchymal stem cells for cardiac repair. Sci. Adv. 2020, 6, eaaz0952. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Kim, T.J.; Kang, L.; Kim, Y.J.; Kang, M.K.; Kim, J.; Ryu, J.H.; Hyeon, T.; Yoon, B.W.; Ko, S.B.; et al. Mesenchymal stem cell-derived magnetic extracellular nanovesicles for targeting and treatment of ischemic stroke. Biomaterials 2020, 243, 119942. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, R.; Naitou, H.; Kunimasa, K.; Ayuzawa, R.; Fujimori, Y.; Ohashi, N.; Kaji, K.; Ohta, T. Proteomic analysis of hypoxia-induced tube breakdown of an in vitro capillary model composed of HUVECs: Potential role of p38-regulated reduction of HSP27. Proteomics 2008, 8, 2897–2906. [Google Scholar] [CrossRef] [PubMed]
- Li, T.F.; Yan, Y.M.; Wang, B.Y.; Qian, H.; Zhang, X.; Shen, L.; Wang, M.; Zhou, Y.; Zhu, W.; Li, W.; et al. Exosomes Derived from Human Umbilical Cord Mesenchymal Stem Cells Alleviate Liver Fibrosis. Stem Cells Dev. 2013, 22, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.M.; He, C. The glial scar in spinal cord injury and repair. Neurosci. Bull. 2013, 29, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Cua, R.C.; Lau, L.W.; Keough, M.B.; Midha, R.; Apte, S.S.; Yong, V.W. Overcoming neurite-inhibitory chondroitin sulfate proteoglycans in the astrocyte matrix. Glia 2013, 61, 972–984. [Google Scholar] [CrossRef]
- Kroner, A.; Greenhalgh, A.D.; Zarruk, J.G.; dos Santos, R.P.; Gaestel, M.; David, S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 2014, 83, 1098–1116. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cao, K.; Sun, X.; Chen, Y.X.; Duan, Z.X.; Sun, L.; Guo, L.; Bai, P.; Sun, D.M.; Fan, J.Q.; et al. Macrophages in spinal cord injury: Phenotypic and functional change from exposure to myelin debris. Glia 2015, 63, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Shin, T.; Ahn, M.; Moon, C.; Kim, S.; Sim, K.B. Alternatively activated macrophages in spinal cord injury and remission: Another mechanism for repair? Mol. Neurobiol. 2013, 47, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Greenhalgh, A.D.; Kroner, A. Macrophage and microglial plasticity in the injured spinal cord. Neuroscience 2015, 307, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Bethea, J.R.; Castro, M.; Keane, R.W.; Lee, T.T.; Dietrich, W.D.; Yezierski, R.P. Traumatic spinal cord injury induces nuclear factor-kappa B activation. J. Neurosci. 1998, 18, 251–3260. [Google Scholar] [CrossRef]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.H.; Hu, C.M.J.; Luk, B.T.; Gao, W.W.; Copp, J.A.; Tai, Y.Y.; O′Connor, D.E.; Zhang, L.F. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014, 14, 2181–2188. [Google Scholar] [CrossRef]
- Tang, J.N.; Su, T.; Huang, K.; Dinh, P.U.; Wang, Z.G.; Vandergriff, A.; Hensley, M.T.; Cores, J.; Allen, T.; Li, T.S.; et al. Targeted repair of heart injury by stem cells fused with platelet nanovesicles. Nat. Biomed. Eng. 2018, 2, 17–26. [Google Scholar] [CrossRef]
- Basso, D.M.; Fisher, L.C.; Anderson, A.J.; Jakeman, L.B.; McTigue, D.M.; Popovich, P.G. Basso mouse scale for locomotion detects differences in recovery after spinal cord in ury in five common mouse strains. J. Neurotrauma 2006, 23, 635–659. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-R.; Kyung, J.W.; Kumar, H.; Kwon, S.P.; Song, S.Y.; Han, I.-B.; Kim, B.-S. Targeted Delivery of Mesenchymal Stem Cell-Derived Nanovesicles for Spinal Cord Injury Treatment. Int. J. Mol. Sci. 2020, 21, 4185. https://doi.org/10.3390/ijms21114185
Lee J-R, Kyung JW, Kumar H, Kwon SP, Song SY, Han I-B, Kim B-S. Targeted Delivery of Mesenchymal Stem Cell-Derived Nanovesicles for Spinal Cord Injury Treatment. International Journal of Molecular Sciences. 2020; 21(11):4185. https://doi.org/10.3390/ijms21114185
Chicago/Turabian StyleLee, Ju-Ro, Jae Won Kyung, Hemant Kumar, Sung Pil Kwon, Seuk Young Song, In-Bo Han, and Byung-Soo Kim. 2020. "Targeted Delivery of Mesenchymal Stem Cell-Derived Nanovesicles for Spinal Cord Injury Treatment" International Journal of Molecular Sciences 21, no. 11: 4185. https://doi.org/10.3390/ijms21114185
APA StyleLee, J. -R., Kyung, J. W., Kumar, H., Kwon, S. P., Song, S. Y., Han, I. -B., & Kim, B. -S. (2020). Targeted Delivery of Mesenchymal Stem Cell-Derived Nanovesicles for Spinal Cord Injury Treatment. International Journal of Molecular Sciences, 21(11), 4185. https://doi.org/10.3390/ijms21114185