1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
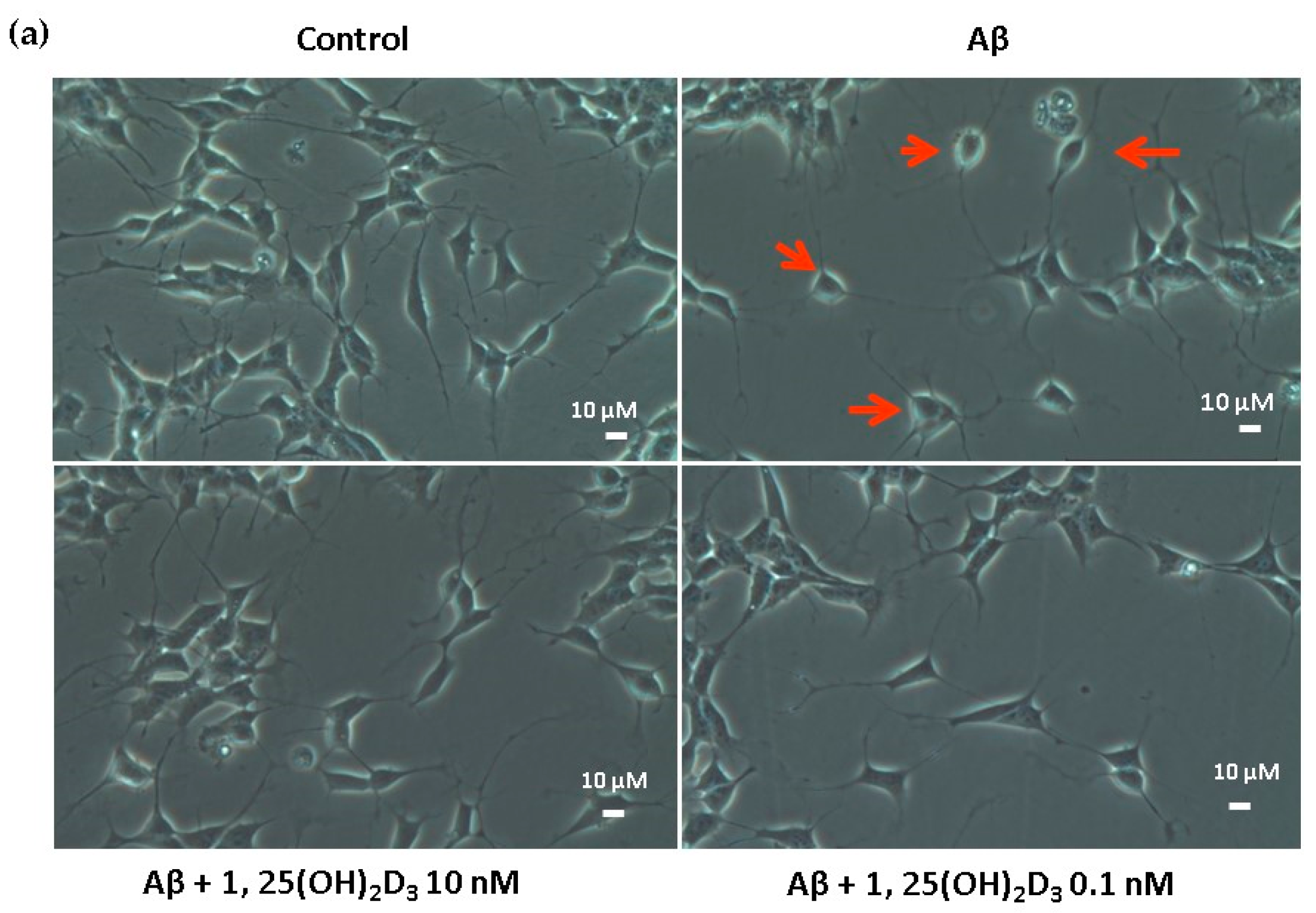
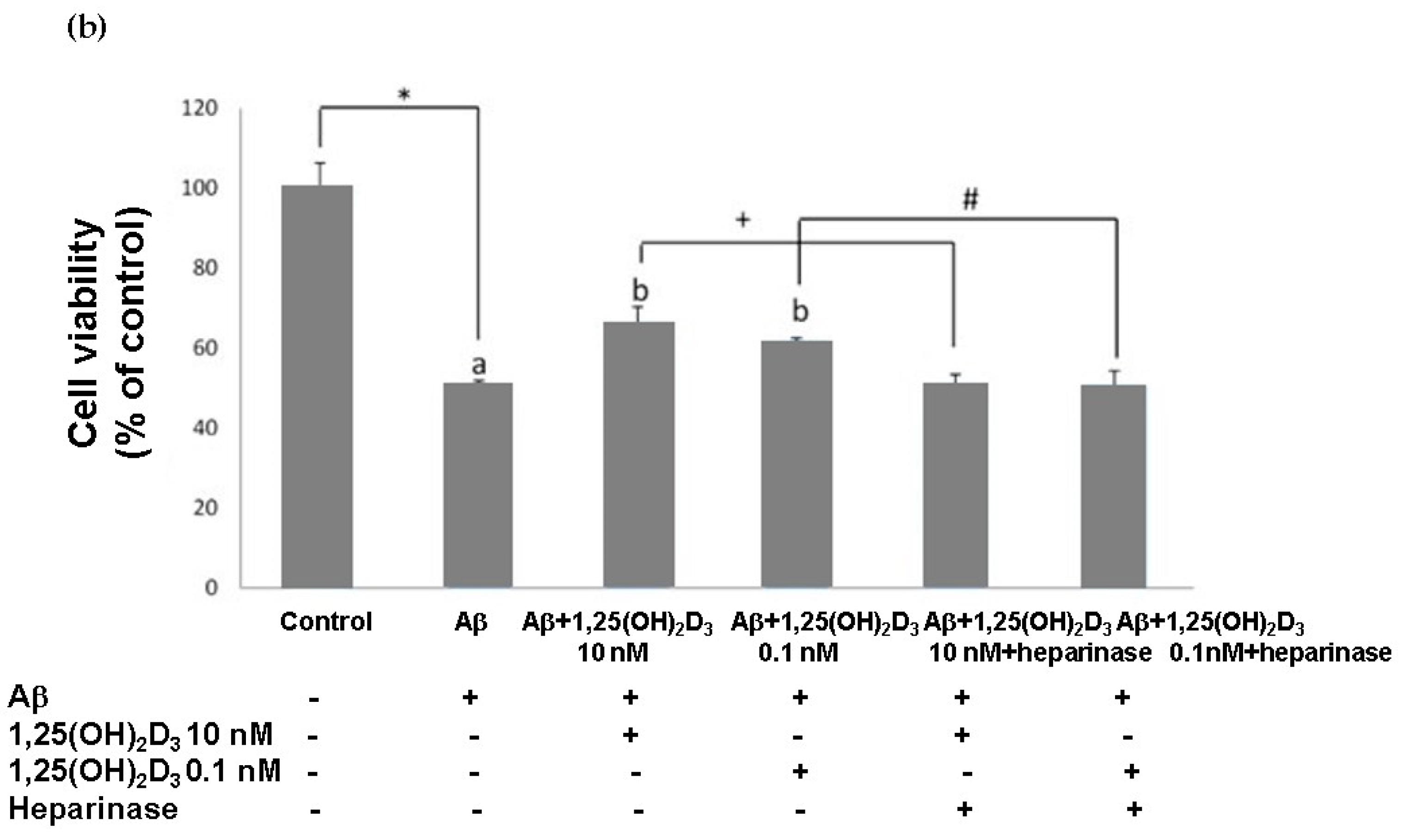
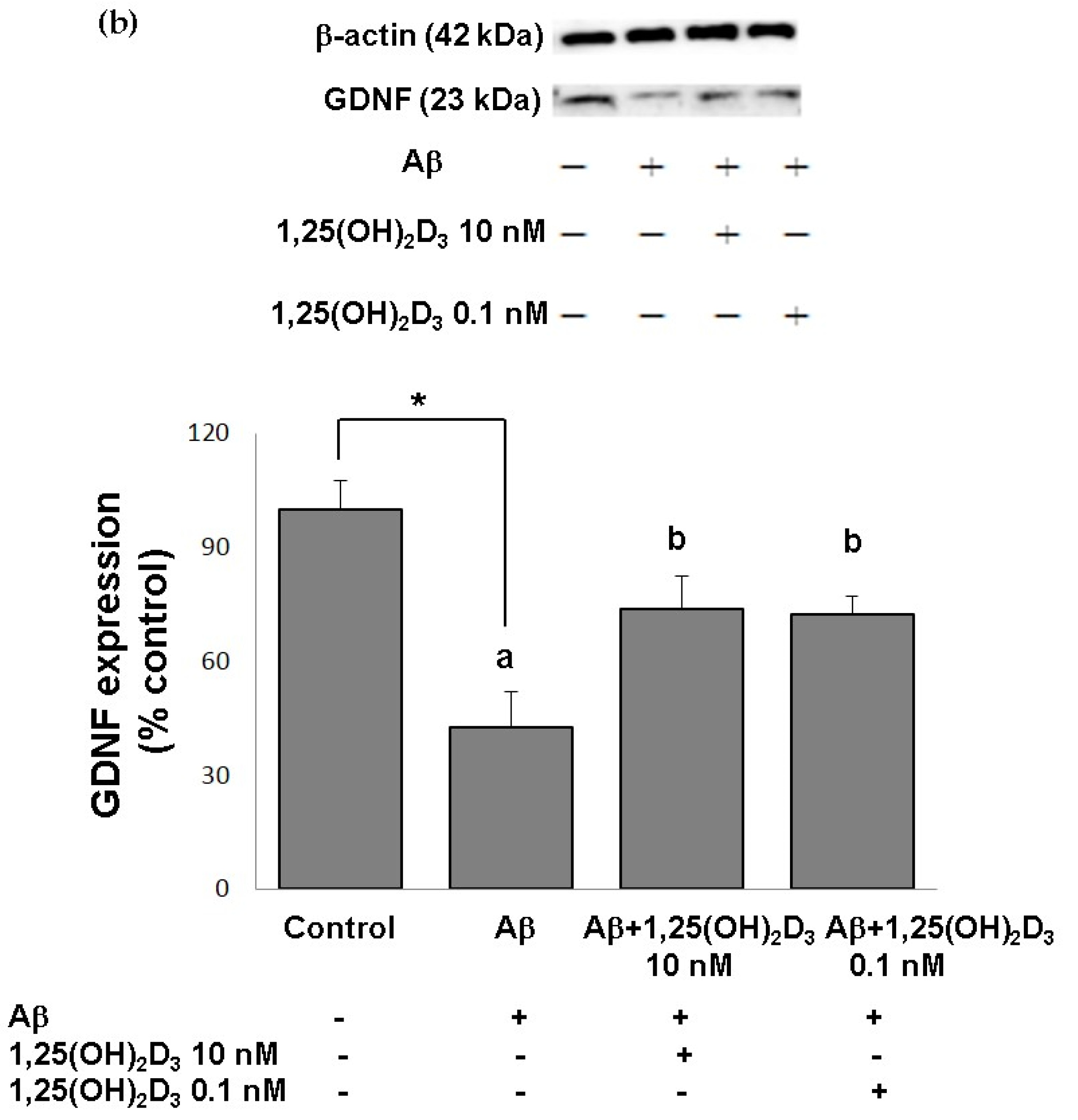
2.1. Effects of 1,25(OH)2D3 on Cell Morphology, Cell Viability, and Protein Expression of VDR and GDNF After Aβ(25-35) Treatment
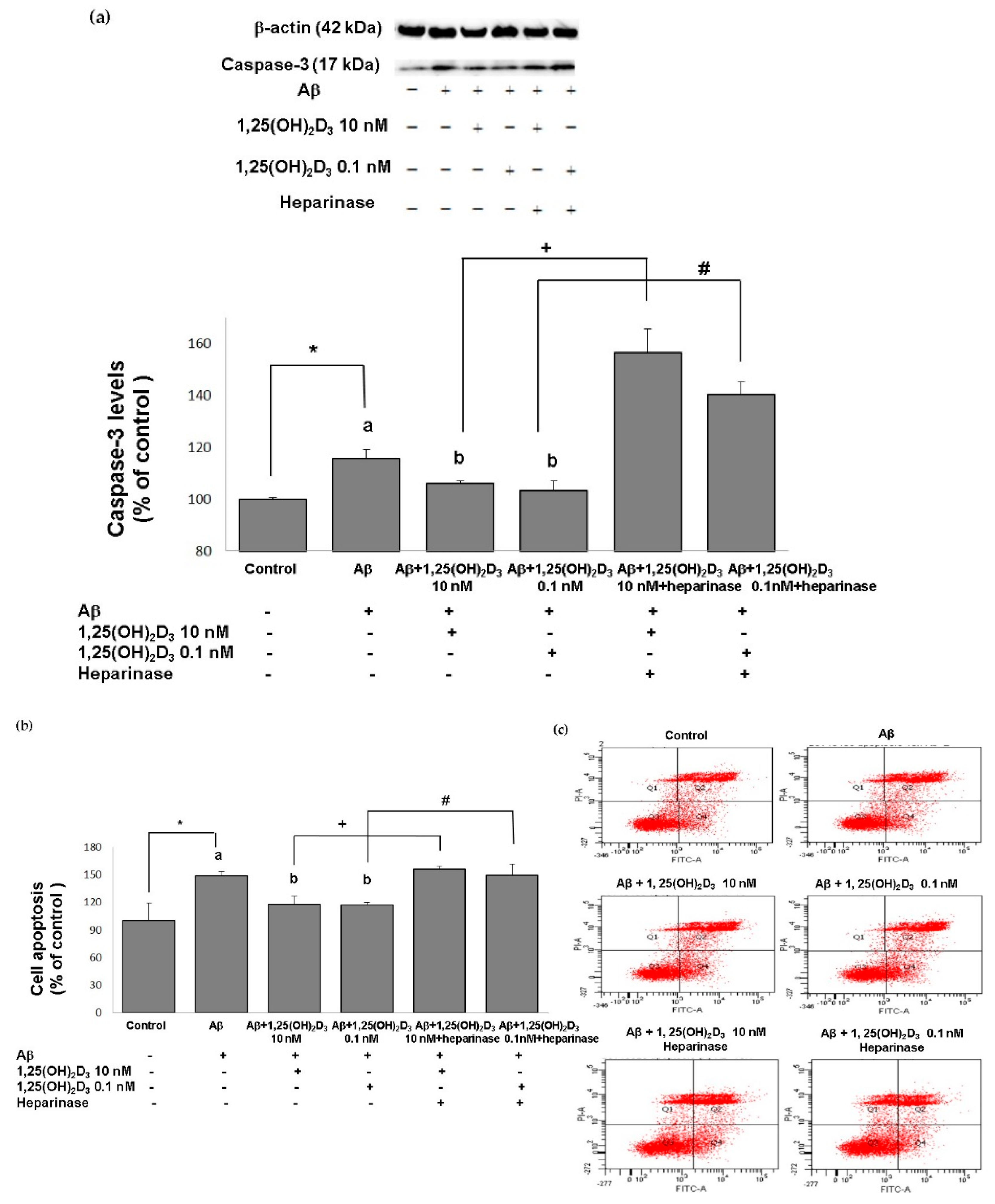
2.2. Effects of 1,25(OH)2D3 on Activating Caspase-3 and Cell Apoptosis after Aβ(25-35) Treatment
2.3. Effects of 1,25(OH)2D3 on Intracellular ROS after Aβ(25-35) Treatment
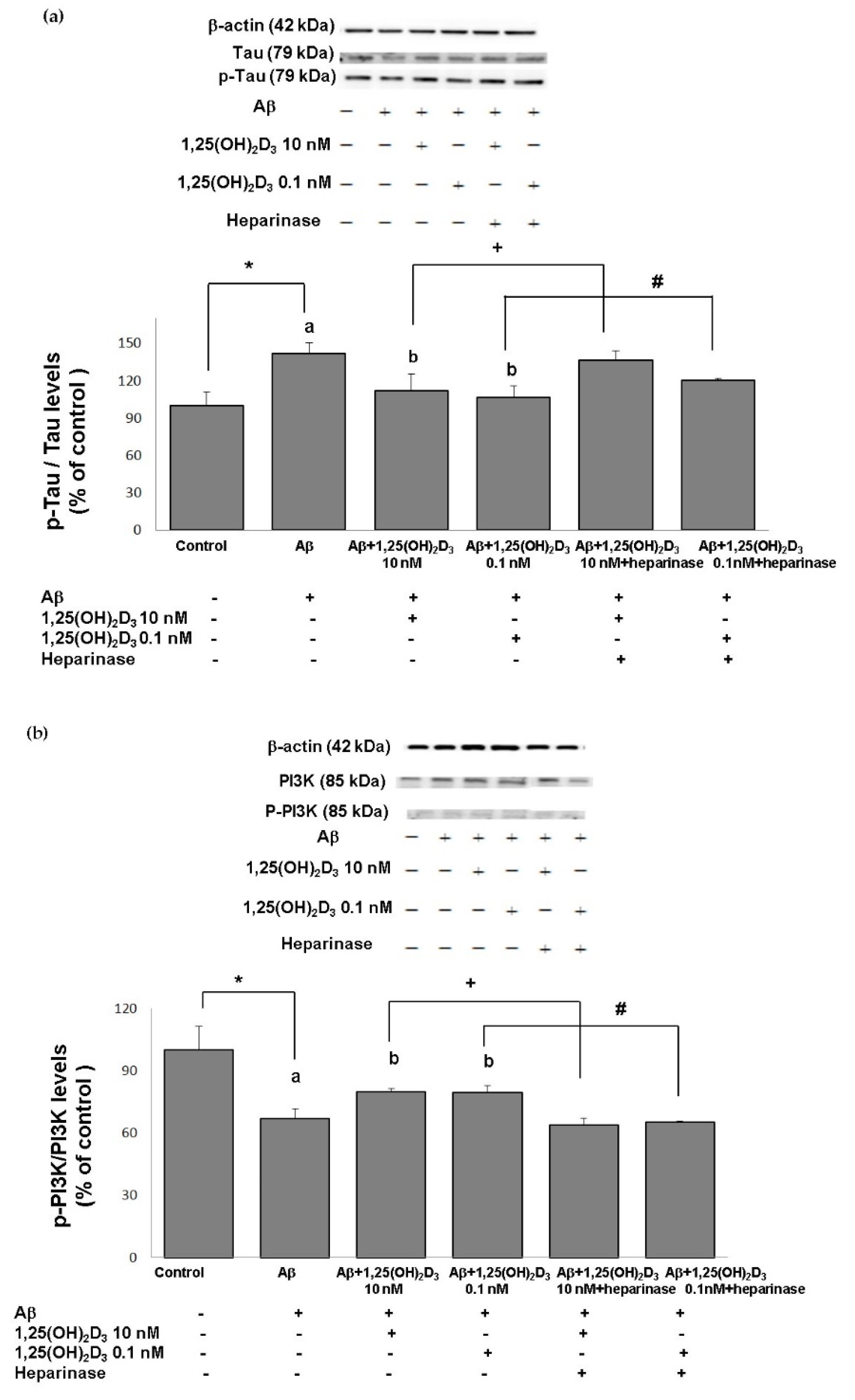
2.4. Effects of 1,25(OH)2D3 on the p-Tau/Tau Ratio after Aβ(25-35) Treatment
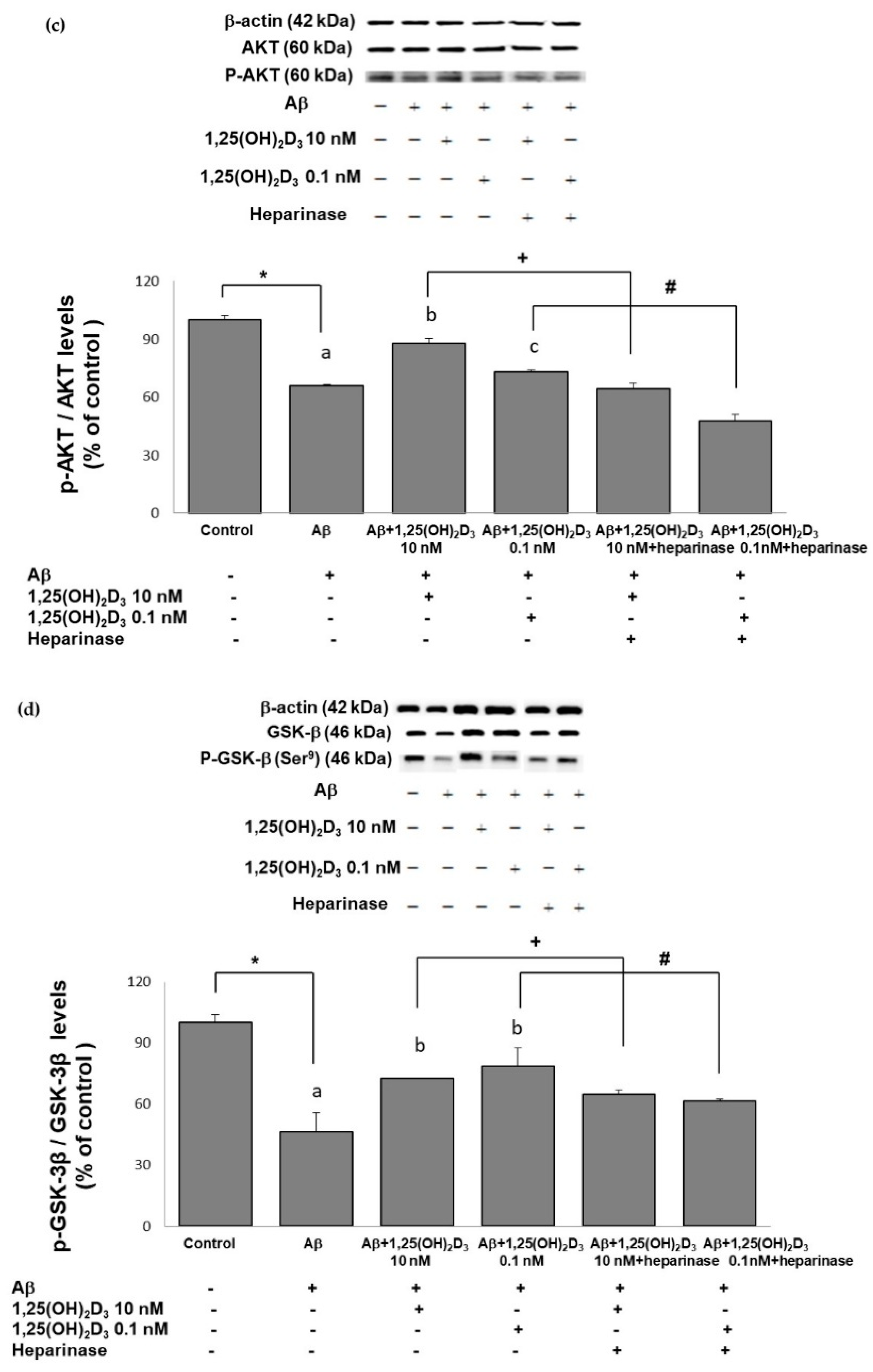
2.5. Effects of 1,25(OH)2D3 on the p-PI3K/PI3K, p-Akt/Akt, and p-GSK-3β (Ser9)/GSK-3β Ratios after Aβ(25-35) Treatment
3. Discussion
4. Materials and Methods
4.1. Aβ(25-35) and 1,25(OH)2D3 Preparations
4.2. Cell Culture Preparation
4.3. Cell Morphology
4.4. Cell Viability Analysis
4.5. Intracellular ROS Analysis
4.6. Protein Extraction and Quantification
4.7. Western Blot Analysis
4.8. Apoptotic Cell Analysis
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| 1,25(OH)2D3 | 1α,25-dihydroxyvitamin D3 |
| VDR | Vitamin D receptor |
| ROS | Reactive oxygen species |
| GDNF | Glial cell line-derived neurotrophic factor |
| PI3K | Phosphoinositide 3-kinase |
| Akt | Protein kinase B |
| GSK-3β | Glycogen synthase kinase-3β |
| APP | Amyloid protein precursor |
| DCFH-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| DMEM | Dulbecco’s modified Eagle medium |
| DCF | Dichlorofluorescein |
| MTT | 3-[4,5-cimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide |
| PBS | Phosphate-buffered saline |
| DMSO | Dimethyl sulfoxide |
| SDS | Sodium dodecyl sulfate |
| TBS | Tris-buffered saline |
| TBST | Tris-buffered saline containing 0.1% Tween-20 |
| ECL | Enhanced chemiluminescence |
| IgG | Immunoglobulin G |
| PI | Propidium iodide |
| ANOVA | Analysis of variance |
| SD | Standard deviation |
References
- Misonou, H.; Morishima-Kawashima, M.; Ihara, Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 2000, 39, 6951–6959. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastian-Serrano, A.; de Diego-Garcia, L.; Diaz-Hernandez, M. The Neurotoxic Role of Extracellular Tau Protein. Int. J. Mol. Sci. 2018, 19, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Jiang, Z.F. Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2009, 16, 15–27. [Google Scholar] [CrossRef]
- Van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13. [Google Scholar] [CrossRef]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Boil. 2000, 130, 184–208. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 2005, 1703, 149–156. [Google Scholar] [CrossRef]
- Varadarajan, S.; Yatin, S.; Kanski, J.; Jahanshahi, F.; Butterfield, D.A. Methionine residue 35 is important in amyloid beta-peptide-associated free radical oxidative stress. Brain Res. Bull. 1999, 50, 133–141. [Google Scholar] [CrossRef]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25-35. Curr. Protein Pept. sci. 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Clementi, M.E.; Marini, S.; Coletta, M.; Orsini, F.; Giardina, B.; Misiti, F. Abeta(31-35) and Abeta(25-35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: Role of the redox state of methionine-35. FEBS Lett. 2005, 579, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Xiong, N.; Huang, C.; Tang, Q.; Hu, B.; Xiang, J.; Li, G. Erythropoietin protects PC12 cells from beta-amyloid(25-35)-induced apoptosis via PI3K/Akt signaling pathway. Neuropharmacology 2009, 56, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Hetman, M.; Cavanaugh, J.E.; Kimelman, D.; Xia, Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 2567–2574. [Google Scholar] [CrossRef] [Green Version]
- Anjum, I.; Jaffery, S.S.; Fayyaz, M.; Samoo, Z.; Anjum, S. The Role of Vitamin D in Brain Health: A Mini Literature Review. Cureus 2018, 10, e2960. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Shah, J. Role of Vitamin D in Amyloid clearance via LRP-1 upregulation in Alzheimer’s disease: A potential therapeutic target? J. Chem. Neuroanat. 2017, 85, 36–42. [Google Scholar] [CrossRef]
- Annweiler, C.; Rolland, Y.; Schott, A.M.; Blain, H.; Vellas, B.; Herrmann, F.R.; Beauchet, O. Higher vitamin D dietary intake is associated with lower risk of alzheimer’s disease: A 7-year follow-up. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2012, 67, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Briones, T.L.; Darwish, H. Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J. Neuroinflamm. 2012, 9, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annweiler, C. Vitamin D in dementia prevention. Ann. N. Y. Acad. Sci. 2016, 1367, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Hsieh, J.C.; Thompson, P.D.; Selznick, S.H.; Dominguez, C.E.; Jurutka, P.W. The nuclear vitamin D receptor: Biological and molecular regulatory properties revealed. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1998, 13, 325–349. [Google Scholar] [CrossRef] [PubMed]
- Buell, J.S.; Dawson-Hughes, B. Vitamin D and neurocognitive dysfunction: Preventing “D”ecline? Mol. Asp. Med. 2008, 29, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissou, M.F.; Guttin, A.; Zenga, C.; Berger, F.; Issartel, J.P.; Wion, D. Additional clues for a protective role of vitamin D in neurodegenerative diseases: 1,25-dihydroxyvitamin D3 triggers an anti-inflammatory response in brain pericytes. J. Alzheimer’s Dis. JAD 2014, 42, 789–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibi, M.; Sawada, H.; Nakanishi, M.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Protective effects of 1 alpha,25-(OH)(2)D(3) against the neurotoxicity of glutamate and reactive oxygen species in mesencephalic culture. Neuropharmacology 2001, 40, 761–771. [Google Scholar] [CrossRef]
- Masoumi, A.; Goldenson, B.; Ghirmai, S.; Avagyan, H.; Zaghi, J.; Abel, K.; Zheng, X.; Espinosa-Jeffrey, A.; Mahanian, M.; Liu, P.T.; et al. 1alpha,25-dihydroxyvitamin D3 interacts with curcuminoids to stimulate amyloid-beta clearance by macrophages of Alzheimer’s disease patients. J. Alzheimer’s Dis. JAD 2009, 17, 703–717. [Google Scholar] [CrossRef]
- Sun, C.; Ou, X.; Farley, J.M.; Stockmeier, C.; Bigler, S.; Brinton, R.D.; Wang, J.M. Allopregnanolone increases the number of dopaminergic neurons in substantia nigra of a triple transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 473–480. [Google Scholar]
- Airaksinen, M.S.; Titievsky, A.; Saarma, M. GDNF family neurotrophic factor signaling: Four masters, one servant? Mol. Cell. Neurosci. 1999, 13, 313–325. [Google Scholar] [CrossRef]
- Chen, P.S.; Peng, G.S.; Li, G.; Yang, S.; Wu, X.; Wang, C.C.; Wilson, B.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatr. 2006, 11, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Gordon, T. The role of neurotrophic factors in nerve regeneration. Neurosurg. Focus 2009, 26, E3. [Google Scholar] [CrossRef]
- Duarte, E.P.; Curcio, M.; Canzoniero, L.M.; Duarte, C.B. Neuroprotection by GDNF in the ischemic brain. Growth Factors 2012, 30, 242–257. [Google Scholar] [CrossRef]
- Eyles, D.; Brown, J.; Mackay-Sim, A.; McGrath, J.; Feron, F. Vitamin D 3 and brain development. Neuroscience 2003, 118, 641–653. [Google Scholar] [CrossRef]
- Straten, G.; Eschweiler, G.W.; Maetzler, W.; Laske, C.; Leyhe, T. Glial cell-line derived neurotrophic factor (GDNF) concentrations in cerebrospinal fluid and serum of patients with early Alzheimer’s disease and normal controls. J. Alzheimer’s Dis. JAD 2009, 18, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Lefter, R.; Serban, I.L.; Stefanescu, C.; Chirita, R. The oxidative stress hypothesis in Alzheimer’s disease. Psychiatr. Danub. 2013, 25, 401–409. [Google Scholar]
- Giraldo, E.; Lloret, A.; Fuchsberger, T.; Vina, J. Abeta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: Protective role of vitamin E. Redox Boil. 2014, 2, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Bissoyi, A.; Kashyap, M.P.; Patra, P.K.; Rizvi, S.I. Autophagy Activation Alleviates Amyloid-beta-Induced Oxidative Stress, Apoptosis and Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells. Neurotox. Res. 2017, 32, 351–361. [Google Scholar] [CrossRef]
- Leong, Y.Q.; Ng, K.Y.; Chye, S.M.; Ling, A.P.K.; Koh, R.Y. Mechanisms of action of amyloid-beta and its precursor protein in neuronal cell death. Metab. Brain Dis. 2020, 35, 11–30. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, F.; Wang, K.X.; Zhou, Y.Z.; Du, G.H.; Qin, X.M. Baicalein protects PC12 cells from Abeta25-35-induced cytotoxicity via inhibition of apoptosis and metabolic disorders. Life Sci. 2020, 248, 117471. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, Y.; Wang, H.; Zhao, L.; Ma, Z.; Li, T.; Liu, J.; Sun, M.; Jian, Y.; Yao, L.; et al. Edaravone reduces Abeta-induced oxidative damage in SH-SY5Y cells by activating the Nrf2/ARE signaling pathway. Life Sci. 2019, 221, 259–266. [Google Scholar] [CrossRef]
- Mohamed, N.V.; Herrou, T.; Plouffe, V.; Piperno, N.; Leclerc, N. Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur. J. Neurosci. 2013, 37, 1939–1948. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.K.; Weindruch, R.; Prolla, T.A. Gene-expression profile of the ageing brain in mice. Nat. Genet. 2000, 25, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Ghribi, O.; Herman, M.M.; Pramoonjago, P.; Spaulding, N.K.; Savory, J. GDNF regulates the A beta-induced endoplasmic reticulum stress response in rabbit hippocampus by inhibiting the activation of gadd 153 and the JNK and ERK kinases. Neurobiol. Dis. 2004, 16, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Alberghina, L.; Colangelo, A. The modular systems biology approach to investigate the control of apoptosis in Alzheimer’s disease neurodegeneration. BMC Neurosci. 2006, 7, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, X. Antioxidant therapies for Alzheimer’s disease. Oxidative Med. Cell. Longev. 2012, 2012, 472932. [Google Scholar] [CrossRef] [Green Version]
- Molinari, C.; Morsanuto, V.; Ghirlanda, S.; Ruga, S.; Notte, F.; Gaetano, L.; Uberti, F. Role of Combined Lipoic Acid and Vitamin D3 on Astrocytes as a Way to Prevent Brain Ageing by Induced Oxidative Stress and Iron Accumulation. Oxidative Med. Cell. Longev. 2019, 2019, 2843121. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Khemka, V.K.; Ganguly, A.; Roy, D.; Ganguly, U.; Chakrabarti, S. Vitamin D and Alzheimer’s Disease: Neurocognition to Therapeutics. Int. J. Alzheimer’s Dis. 2015, 2015, 192747. [Google Scholar] [CrossRef] [Green Version]
- Orme, R.P.; Bhangal, M.S.; Fricker, R.A. Calcitriol Imparts Neuroprotection In Vitro to Midbrain Dopaminergic Neurons by Upregulating GDNF Expression. PLoS ONE 2013, 8, e62040. [Google Scholar] [CrossRef] [Green Version]
- Naveilhan, P.; Neveu, I.; Wion, D.; Brachet, P. 1,25-Dihydroxyvitamin D3, an inducer of glial cell line-derived neurotrophic factor. Neuroreport 1996, 7, 2171–2175. [Google Scholar] [CrossRef]
- Celli, A.; Treves, C.; Stio, M. Vitamin D receptor in SH-SY5Y human neuroblastoma cells and effect of 1,25-dihydroxyvitamin D3 on cellular proliferation. Neurochem. Int. 1999, 34, 117–124. [Google Scholar] [CrossRef]
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New clues about vitamin D functions in the nervous system. Trends Endocrinol. Metab. TEM 2002, 13, 100–105. [Google Scholar] [CrossRef]
- Dursun, E.; Gezen-Ak, D.; Yilmazer, S. A novel perspective for Alzheimer’s disease: Vitamin D receptor suppression by amyloid-beta and preventing the amyloid-beta induced alterations by vitamin D in cortical neurons. J. Alzheimer’s Dis. JAD 2011, 23, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Neveu, I.; Naveilhan, P.; Baudet, C.; Wion, D.; De Luca, H.F.; Brachet, P. 1, 25-dihydroxyvitamin D 3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Mol. Brain Res. 1994, 24, 70–76. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Wu, J.-N.; Cherng, T.-L.; Hoffer, B.J.; Chen, H.-H.; Borlongan, C.V.; Wang, Y. Vitamin D 3 attenuates 6-hydroxydopamine-induced neurotoxicity in rats. Brain Res. 2001, 904, 67–75. [Google Scholar] [CrossRef]
- Pertile, R.A.N.; Cui, X.; Hammond, L.; Eyles, D.W. Vitamin D regulation of GDNF/Ret signaling in dopaminergic neurons. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Kitagishi, Y.; Kobayashi, M.; Kikuta, K.; Matsuda, S. Roles of PI3K/AKT/GSK3/mTOR pathway in cell signaling of mental illnesses. Depress. Res. Treat. 2012, 2012, 752563. [Google Scholar] [CrossRef] [Green Version]
- Jolivalt, C.; Lee, C.; Beiswenger, K.; Smith, J.; Orlov, M.; Torrance, M.; Masliah, E. Defective insulin signaling pathway and increased GSK-3 activity in the brain of diabetic mice: Parallels with Alzheimer’s disease and correction by insulin. J. Neurosci. Res. 2008, 86, 3265. [Google Scholar] [CrossRef] [Green Version]
- Villegas, S.N.; Njaine, B.; Linden, R.; Carri, N.G. Glial-derived neurotrophic factor (GDNF) prevents ethanol (EtOH) induced B92 glial cell death by both PI3K/AKT and MEK/ERK signaling pathways. Brain Res. Bull. 2006, 71, 116–126. [Google Scholar] [CrossRef]
- Srinivasan, S.; Anitha, M.; Mwangi, S.; Heuckeroth, R.O. Enteric neuroblasts require the phosphatidylinositol 3-kinase/Akt/Forkhead pathway for GDNF-stimulated survival. Mol. Cell. Neurosci. 2005, 29, 107–119. [Google Scholar] [CrossRef]
- Mograbi, B.; Bocciardi, R.; Bourget, I.; Busca, R.; Rochet, N.; Farahi-Far, D.; Juhel, T.; Rossi, B. Glial cell line-derived neurotrophic factor-stimulated phosphatidylinositol 3-kinase and Akt activities exert opposing effects on the ERK pathway: Importance for the rescue of neuroectodermic cells. J. Boil. Chem. 2001, 276, 45307–45319. [Google Scholar] [CrossRef] [Green Version]
- Besset, V.; Scott, R.P.; Ibanez, C.F. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J. Boil. Chem. 2000, 275, 39159–39166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Mwangi, S.; Anitha, M.; Fu, H.; Sitaraman, S.V.; Srinivasan, S. Glial cell line-derived neurotrophic factor-mediated enteric neuronal survival involves glycogen synthase kinase-3beta phosphorylation and coupling with 14-3-3. Neuroscience 2006, 143, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.J.; Howlett, D.R.; Francis, P.T.; Williams, R.J. Abeta (1-42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol. Aging 2008, 29, 992–1001. [Google Scholar] [CrossRef]
- Schindowski, K.; Belarbi, K.; Buee, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7 (Suppl. 1), 43–56. [Google Scholar] [CrossRef]
- Cora, O.N. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Nayak, G.; Cooper, G.M. p53 is a major component of the transcriptional and apoptotic program regulated by PI 3-kinase/Akt/GSK3 signaling. Cell Death Dis. 2012, 3, e400. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-C.; Leu, S.-J.; Chuang, D.-M. Roles of Glycogen Synthase Kinase-3 in Alzheimer’s Disease: From Pathology to Treatment Target. J. Exp. Clin. Med. 2012, 4, 135–139. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Mirshafiey, A. The immunopathogenic role of reactive oxygen species in Alzheimer disease. Iran. J. Allerg. Asthma Immunol. 2012, 11, 203–216. [Google Scholar]
- Kubo, T.; Nishimura, S.; Kumagae, Y.; Kaneko, I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1-40) to truncated and toxic fragments ([D-Ser 26]A beta 25-35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 2002, 70, 474–483. [Google Scholar] [CrossRef]
- Yang, R.; Chen, L.; Wang, H.; Xu, B.; Tomimoto, H.; Chen, L. Anti-amnesic effect of neurosteroid PREGS in Abeta25-35-injected mice through sigma1 receptor- and alpha7nAChR-mediated neuroprotection. Neuropharmacology 2012, 63, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Guan, Y.L.; Zou, L.B.; Gong, Y.X.; Hua, H.M.; Xu, Y.N.; Zhang, H.; Yu, Z.G.; Fan, W.H. Saponins with neuroprotective effects from the roots of Pulsatilla cernua. Molecules 2012, 17, 5520–5531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awale, S.; Tohda, C.; Tezuka, Y.; Miyazaki, M.; Kadota, S. Protective Effects of Rosa damascena and Its Active Constituent on Aβ (25-35)-Induced Neuritic Atrophy. Evid.-Based Complement. Altern. Med. eCAM 2011, 2011, 131042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przybelski, R.J.; Binkley, N.C. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch. Biochem. Biophys. 2007, 460, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, M.; Ittrich, C.; Wiedmann, V.; Knyazev, Y.; Weninger, A.; Riemenschneider, M.; Hartmann, T. New Alzheimer amyloid beta responsive genes identified in human neuroblastoma cells by hierarchical clustering. PLoS ONE 2009, 4, e6779. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-I.; Chang, Y.-C.; Kao, N.-J.; Lee, W.-J.; Cross, T.-W.; Lin, S.-H. 1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 4215. https://doi.org/10.3390/ijms21124215
Lin C-I, Chang Y-C, Kao N-J, Lee W-J, Cross T-W, Lin S-H. 1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells. International Journal of Molecular Sciences. 2020; 21(12):4215. https://doi.org/10.3390/ijms21124215
Chicago/Turabian StyleLin, Ching-I, Yi-Chen Chang, Ning-Jo Kao, Wei-Ju Lee, Tzu-Wen Cross, and Shyh-Hsiang Lin. 2020. "1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells" International Journal of Molecular Sciences 21, no. 12: 4215. https://doi.org/10.3390/ijms21124215
APA StyleLin, C. -I., Chang, Y. -C., Kao, N. -J., Lee, W. -J., Cross, T. -W., & Lin, S. -H. (2020). 1,25(OH)2D3 Alleviates Aβ(25-35)-Induced Tau Hyperphosphorylation, Excessive Reactive Oxygen Species, and Apoptosis Through Interplay with Glial Cell Line-Derived Neurotrophic Factor Signaling in SH-SY5Y Cells. International Journal of Molecular Sciences, 21(12), 4215. https://doi.org/10.3390/ijms21124215