An Inhibitor of the Sodium–Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
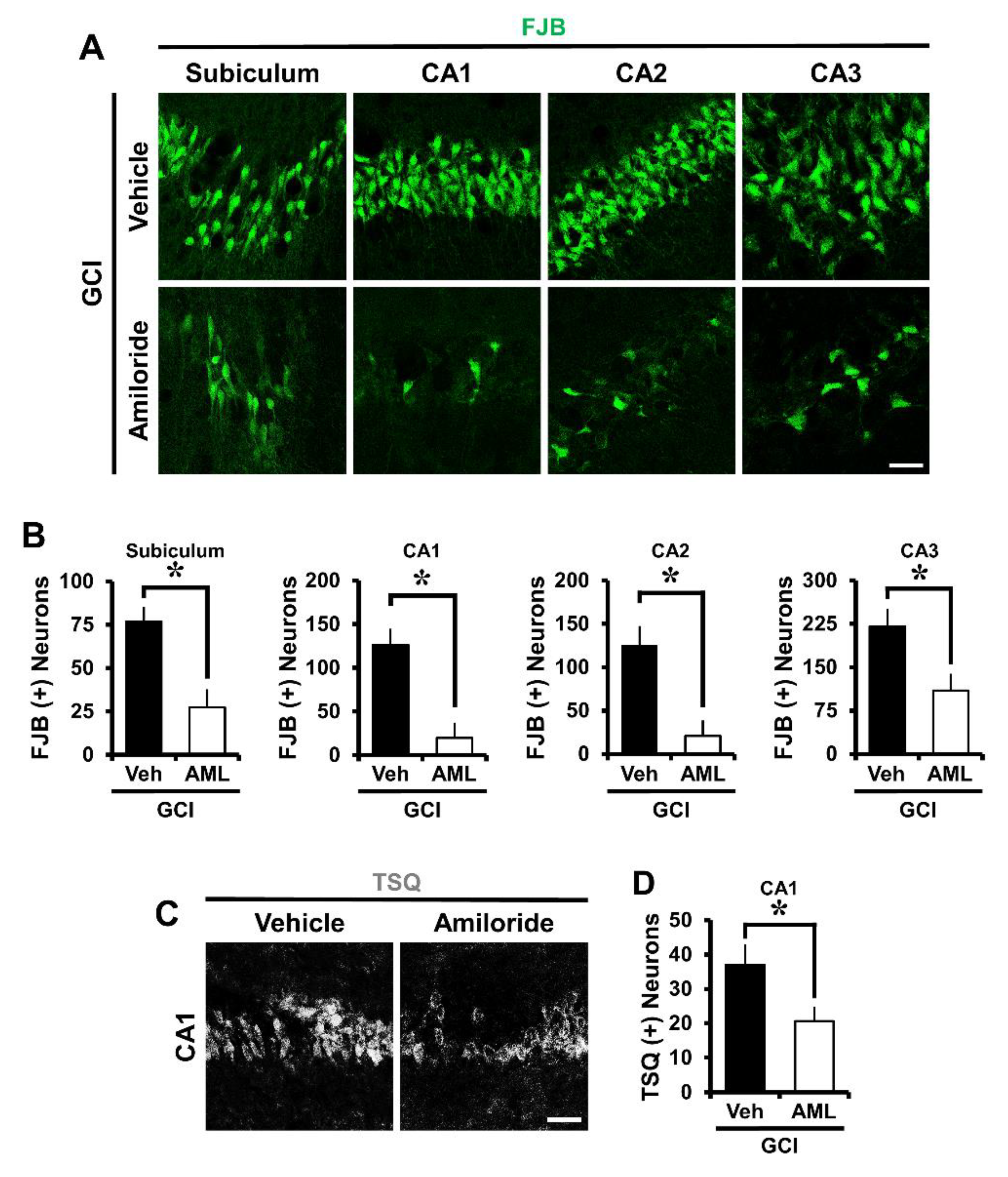
2.1. Amiloride Reduced Global Cerebral Ischemia-Induced Hippocampus Neuronal Death after 24 Hour Post-Insult
2.2. Amiloride Reduced Global Cerebral Ischemia-Induced Hippocampal Zinc Accumulation after 24 Hour Post-Insult
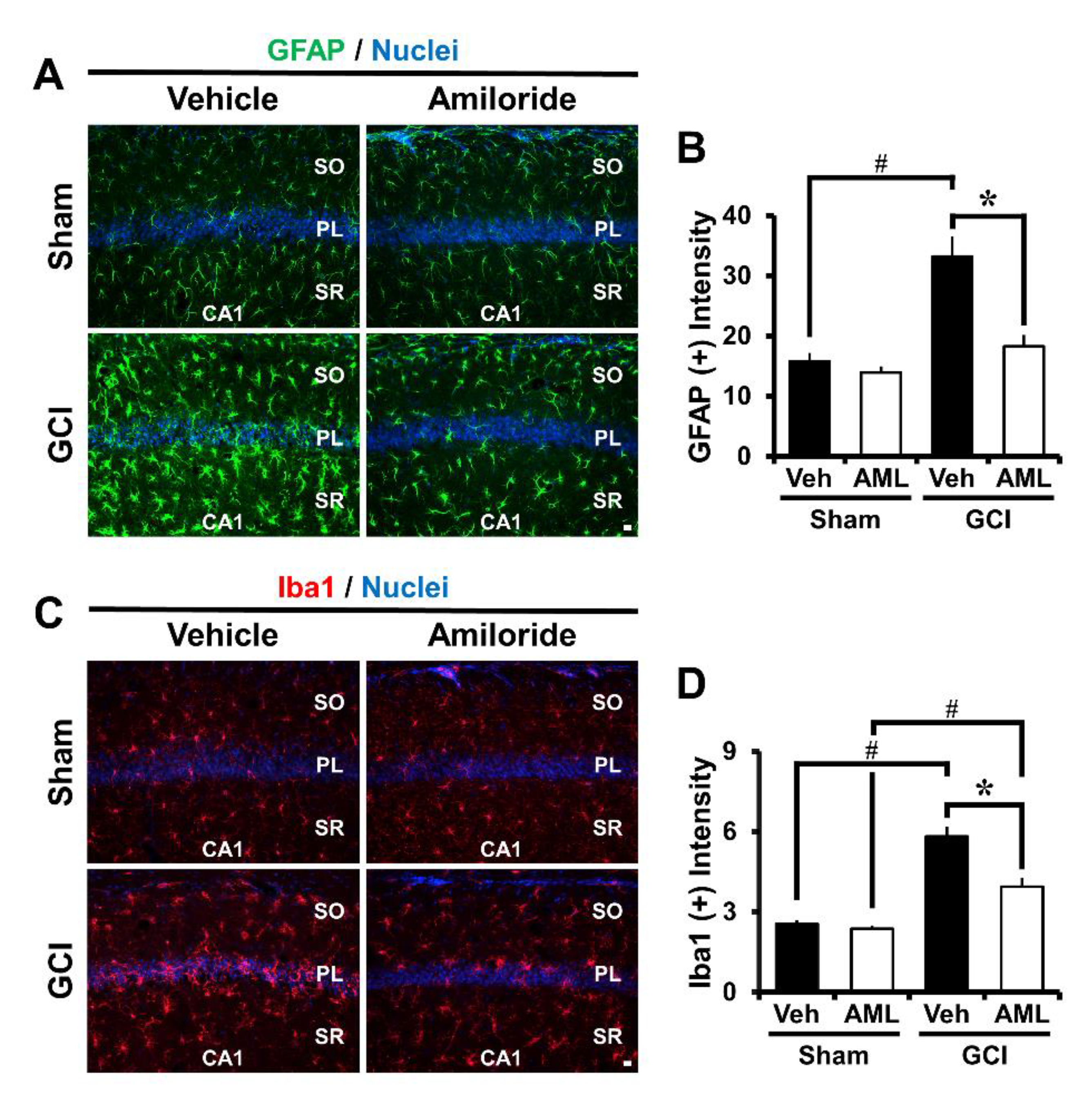
2.3. Amiloride Reduced Global Cerebral Ischemia-Induced Astrocyte and Microglial Activation 24 Hour Post-Insult
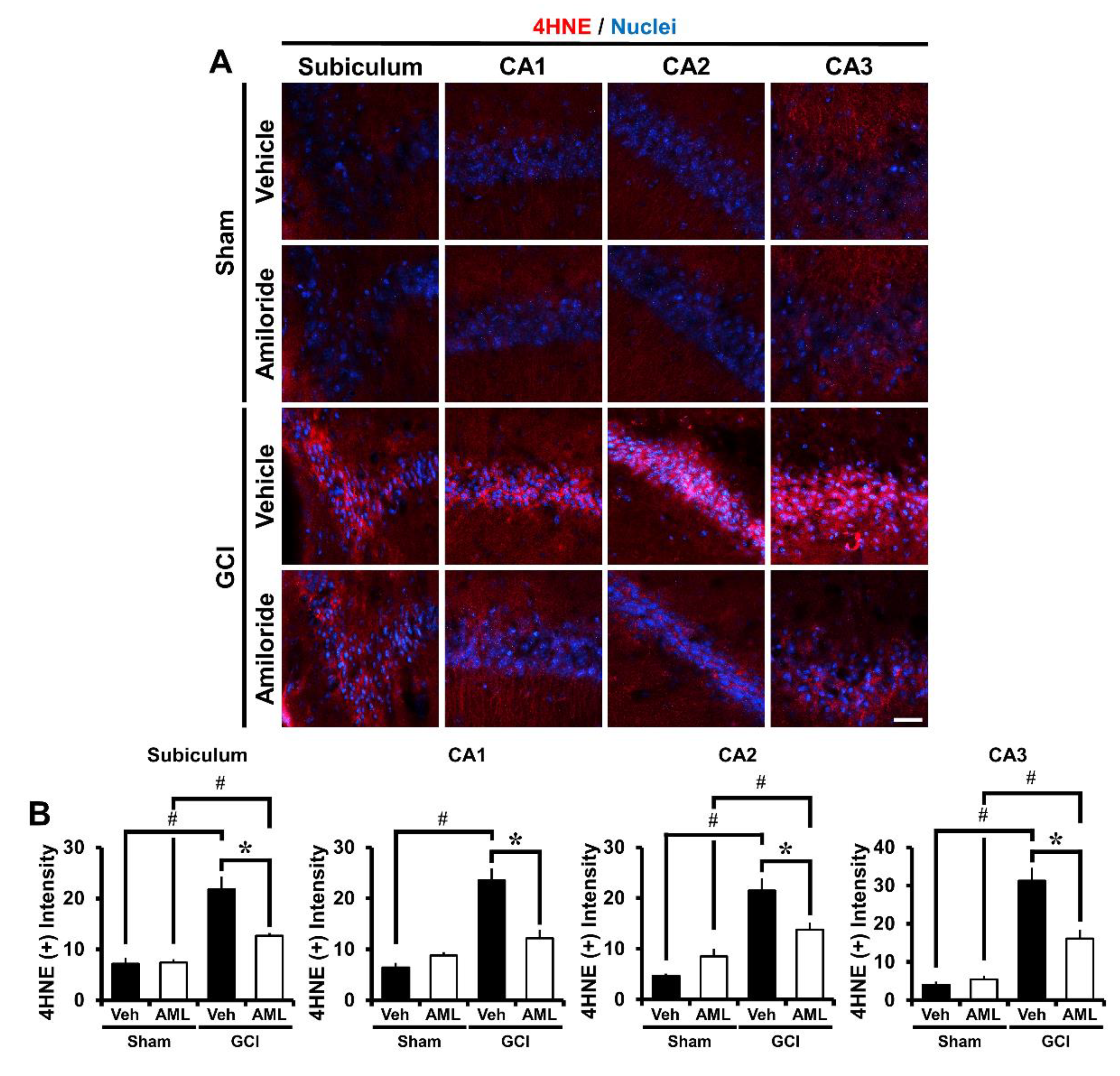
2.4. Amiloride Reduces Global Cerebral Ischemia-Induced Oxidative Damage after 24 Hour Post-Insult
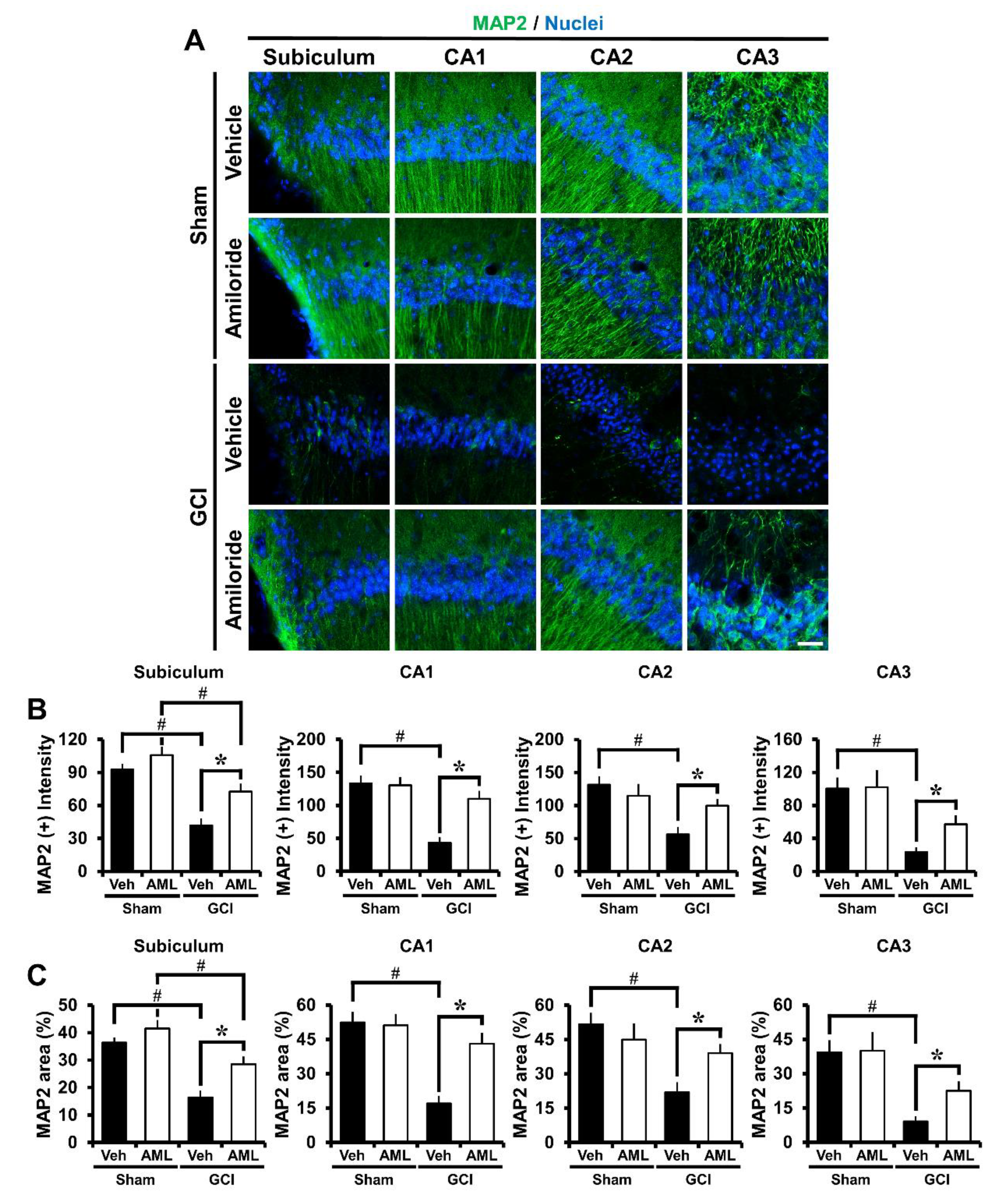
2.5. Amiloride Reduced Global Cerebral Ischemia-Induced Microtubule Damage after 24 Hour Post-Insult
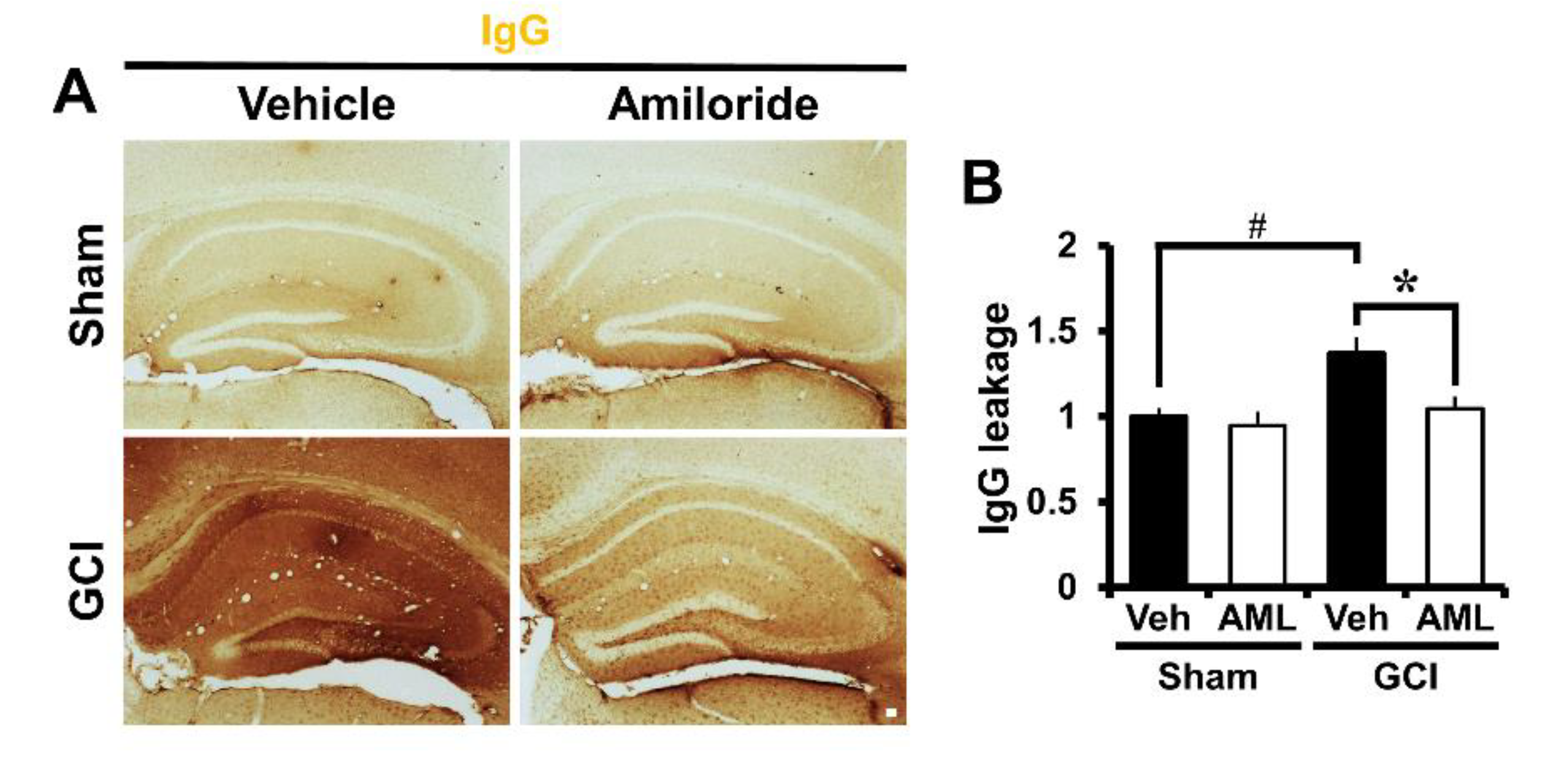
2.6. Amiloride Prevented Global Cerebral Ischemia-Induced Blood–Brain Barrier Disruption after 24 Hour Post-Insult
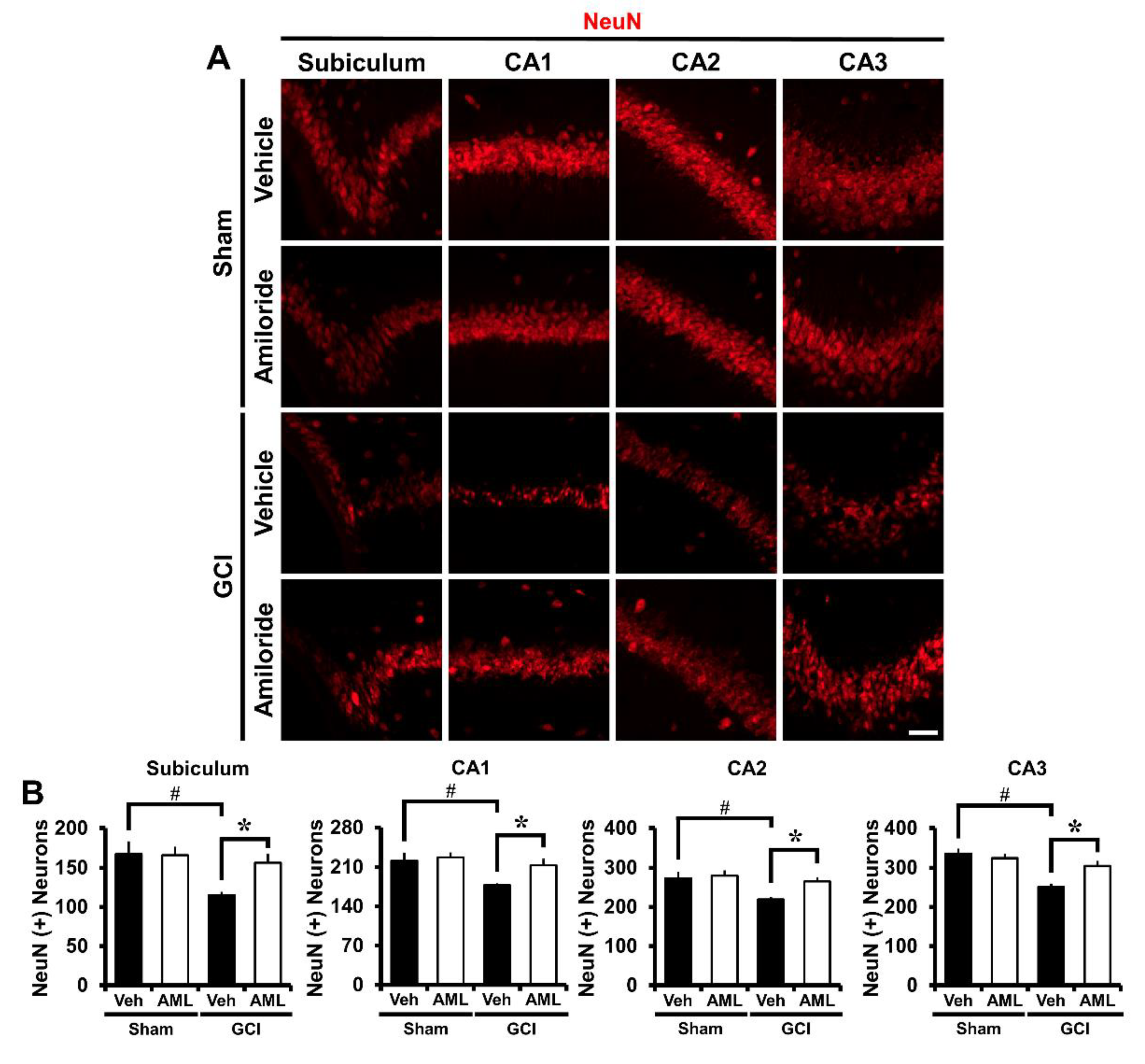
2.7. Amiloride Improves Global Cerebral Ischemia-Induced Survival of Hippocampal Neurons at 3 Days Post-Insult
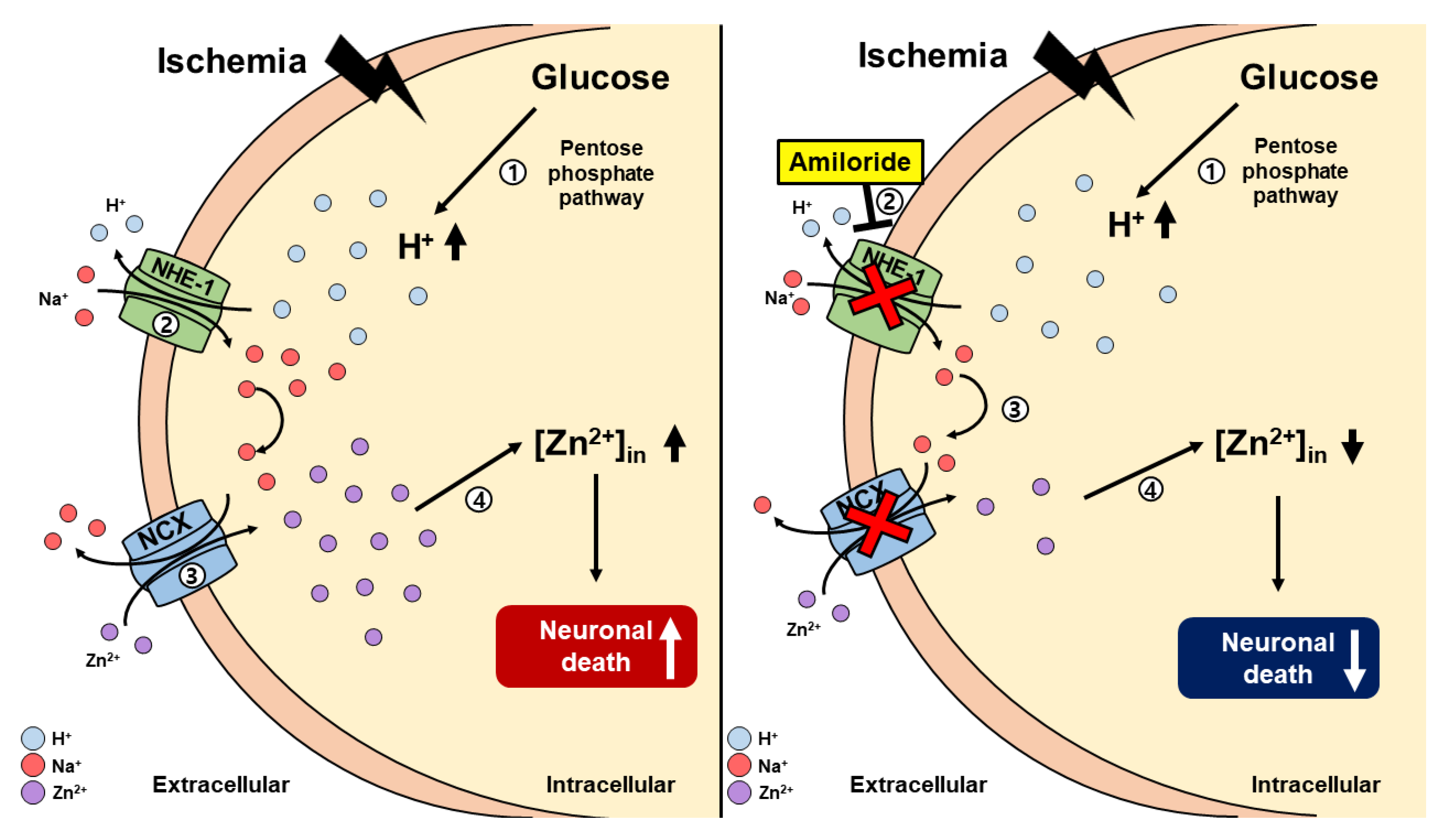
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Animals
4.3. Global Cerebral Ischemia Surgery
4.4. Amiloride Administration
4.5. Brain Sample Preparation
4.6. Confirmation of Hippocampal Neuronal Death
4.7. Confirmation of Hippocampal Zinc Translocation
4.8. Evaluation of Hippocampal Oxidative Stress
4.9. Evaluation of Hippocampal Microtubule Damage
4.10. Evaluation of Hippocampal Astrocytes and Microglia
4.11. Evaluation of BBB Disruption
4.12. Evaluation of Live Hippocampal Neurons
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wan, L.; Cheng, Y.; Luo, Z.; Guo, H.; Zhao, W.; Gu, Q.; Yang, X.; Xu, J.; Bei, W.; Guo, J. Neuroprotection, learning and memory improvement of a standardized extract from Renshen Shouwu against neuronal injury and vascular dementia in rats with brain ischemia. J. Ethnopharmacol. 2015, 165, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.C.; Lee, M.H.H.; Wu, C.Y.C.; Couto, E.S.A.; Possoit, H.E.; Hsieh, T.H.; Minagar, A.; Lin, H.W. Cerebral ischemia and neuroregeneration. Neural Regen. Res. 2018, 13, 373–385. [Google Scholar] [PubMed]
- Hong, D.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Suh, S.W. Carvacrol attenuates hippocampal neuronal death after global cerebral ischemia via inhibition of transient receptor potential melastatin 7. Cells 2018, 7, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Liao, S.J.; Yu, J. Axon guidance factor netrin-1 and its receptors regulate angiogenesis after cerebral ischemia. Neurosci. Bull. 2014, 30, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirnagl, U.; Lindauer, U.; Them, A.; Schreiber, S.; Pfister, H.W.; Koedel, U.; Reszka, R.; Freyer, D.; Villringer, A. Global cerebral ischemia in the rat: Online monitoring of oxygen free radical production using chemiluminescence in vivo. J. Cereb. Blood Flow Metab. 1995, 15, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, B.C.; Sullivan, J.M.; DeGracia, D.J.; O’Neil, B.J.; Neumar, R.W.; Grossman, L.I.; Rafols, J.A.; Krause, G.S. Brain ischemia and reperfusion: Molecular mechanisms of neuronal injury. J. Neurol. Sci. 2000, 179, 1–33. [Google Scholar] [CrossRef]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Klann, E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J. Neurophysiol. 1998, 80, 452–457. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Pan, J.; Konstas, A.A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Rehncrona, S. Brain acidosis. Ann. Emerg. Med. 1985, 14, 770–776. [Google Scholar] [CrossRef]
- Nedergaard, M.; Kraig, R.P.; Tanabe, J.; Pulsinelli, W.A. Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 1991, 260, R581–R588. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K.; Katsura, K.; Kristian, T. Acidosis-related damage. Adv. Neurol. 1996, 71, 209–233. [Google Scholar] [PubMed]
- Cull, R.E. Role of axonal transport in maintaining central synaptic connections. Exp. Brain Res. 1975, 24, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Kodama, Y.; Ohnuma, M.; Okada, S. Zinc transport from the striatum and substantia nigra. Brain Res. Bull. 1998, 47, 103–106. [Google Scholar] [CrossRef]
- Choi, D.W. Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988, 11, 465–469. [Google Scholar] [CrossRef]
- Chuah, M.I.; Tennent, R.; Jacobs, I. Response of olfactory Schwann cells to intranasal zinc sulfate irrigation. J. Neurosci. Res. 1995, 42, 470–478. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of protocatechuic acid (PCA) on global cerebral ischemia-induced hippocampal neuronal death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kwon, L.M.; Sohn, M.; Song, H.K.; Suh, S.W. Zinc chelation reduces hippocampal neurogenesis after pilocarpine-induced seizure. PLoS ONE 2012, 7, e48543. [Google Scholar] [CrossRef]
- Inoue, K.; Branigan, D.; Xiong, Z.G. Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J. Biol. Chem. 2010, 285, 7430–7439. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.Y.; Lee, B.E.; Kim, J.H.; Kim, H.J.; Sohn, M.; Song, H.K.; Chung, T.N.; Suh, S.W. Colchicine induced intraneuronal free zinc accumulation and dentate granule cell degeneration. Metallomics 2014, 6, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ma, E.; Gu, X.Q.; Haddad, G.G. Intracellular pH regulation of CA1 neurons in Na+/H+ isoform 1 mutant mice. J. Clin. Invest. 1999, 104, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, J.; Pouyssegur, J. Hormonal regulation, pharmacology, and membrane sorting of vertebrate Na+/H+ exchanger isoforms. Am. J. Physiol. 1995, 268, C283–C296. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, R.; Matsushita, M.; Kanazawa, H.; Ogihara, S.; Hoekstra, D.; van Ijzendoorn, S.C. The Na+/H+ exchanger NHE6 in the endosomal recycling system is involved in the development of apical bile canalicular surface domains in HepG2 cells. Mol. Biol. Cell 2010, 21, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.; Tanaka, S.; Teko, Y.; Mitsui, K.; Kanazawa, H. Four Na+/H+ exchanger isoforms are distributed to Golgi and post-Golgi compartments and are involved in organelle pH regulation. J. Biol. Chem. 2005, 280, 1561–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleyman, T.R.; Cragoe, E.J., Jr. Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol. 1988, 105, 1–21. [Google Scholar] [CrossRef]
- Hwang, I.K.; Yoo, K.Y.; An, S.J.; Li, H.; Lee, C.H.; Choi, J.H.; Lee, J.Y.; Lee, B.H.; Kim, Y.M.; Kwon, Y.G.; et al. Late expression of Na+/H+ exchanger 1 (NHE1) and neuroprotective effects of NHE inhibitor in the gerbil hippocampal CA1 region induced by transient ischemia. Exp. Neurol. 2008, 212, 314–323. [Google Scholar] [CrossRef]
- Verma, V.; Bali, A.; Singh, N.; Jaggi, A.S. Implications of sodium hydrogen exchangers in various brain diseases. J. Basic Clin. Physiol. Pharmacol. 2015, 26, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.K.; Truong, D.D. Amiloride but not memantine reduces neurodegeneration, seizures and myoclonic jerks in rats with cardiac arrest-induced global cerebral hypoxia and reperfusion. PLoS ONE 2013, 8, e60309. [Google Scholar] [CrossRef] [Green Version]
- Ou-Yang, T.P.; Zhu, G.M.; Ding, Y.X.; Yang, F.; Sun, X.L.; Jiang, W. The effects of amiloride on seizure activity, cognitive deficits and seizure-induced neurogenesis in a novel rat model of febrile seizures. Neurochem. Res. 2016, 41, 933–942. [Google Scholar] [CrossRef]
- Imai, T.; Katoh, H.; Suyama, K.; Kuroiwa, M.; Yanagisawa, S.; Watanabe, M. Amiloride promotes oligodendrocyte survival and remyelination after spinal cord injury in rats. J. Clin. Med. 2018, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, M.; Samman, S. Zinc and redox signaling: Perturbations associated with cardiovascular disease and diabetes mellitus. Antioxid. Redox Signal. 2010, 13, 1549–1573. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.H.; Park, J.B.; Suh, S.W. The effects of sodium dichloroacetate on mitochondrial dysfunction and neuronal death following hypoglycemia-induced injury. Cells 2019, 8, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.M.; Dunn, E.; Oostveen, J.A.; Hall, E.D.; Carter, D.B. Induction of apolipoprotein E mRNA in the hippocampus of the gerbil after transient global ischemia. Brain Res. Mol. Brain Res. 1996, 38, 37–44. [Google Scholar] [CrossRef]
- Hoane, M.R.; Kaplan, S.A.; Ellis, A.L. The effects of nicotinamide on apoptosis and blood-brain barrier breakdown following traumatic brain injury. Brain Res. 2006, 1125, 185–193. [Google Scholar] [CrossRef]
- Tang, X.N.; Berman, A.E.; Swanson, R.A.; Yenari, M.A. Digitally quantifying cerebral hemorrhage using Photoshop and Image J. J. Neurosci. Methods 2010, 190, 240–243. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ward, R.; Dong, G.; Ergul, A.; O’Connor, P. Neurovascular protection in voltage-gated proton channel Hv1 knock-out rats after ischemic stroke: Interaction with Na+/H+ exchanger-1 antagonism. Physiol. Rep. 2019, 7, e14142. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Nguyen, N.; DeGrado, T.R.; Schwaiger, M.; Brosius, F.C., 3rd. Ischemia induces translocation of the insulin-responsive glucose transporter GLUT4 to the plasma membrane of cardiac myocytes. Circulation 1994, 89, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Abel, E.D. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circulation 2001, 103, 2961–2966. [Google Scholar] [CrossRef] [Green Version]
- Young, L.H.; Renfu, Y.; Russell, R.; Hu, X.; Caplan, M.; Ren, J.; Shulman, G.I.; Sinusas, A.J. Low-flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation 1997, 95, 415–422. [Google Scholar] [CrossRef]
- Wang, Y.P.; Zhou, L.S.; Zhao, Y.Z.; Wang, S.W.; Chen, L.L.; Liu, L.X.; Ling, Z.Q.; Hu, F.J.; Sun, Y.P.; Zhang, J.Y.; et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014, 33, 1304–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant. Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Orlowski, J.; Grinstein, S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflügers Arch. 2004, 447, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Avkiran, M. Protection of the ischaemic myocardium by Na+/H+ exchange inhibitors: Potential mechanisms of action. Basic Res. Cardiol. 2001, 96, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Ferrazzano, P.; Shi, Y.; Manhas, N.; Wang, Y.; Hutchinson, B.; Chen, X.; Chanana, V.; Gerdts, J.; Meyerand, M.E.; Sun, D. Inhibiting the Na+/H+ exchanger reduces reperfusion injury: A small animal MRI study. Front. Biosci. (Elite Ed.) 2011, 3, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeves, A.M.; Ennis, I.L. Na+/H+ exchanger and cardiac hypertrophy. Hipertens. Riesgo. Vasc. 2020, 37, 22–32. [Google Scholar] [CrossRef]
- Sensi, S.L.; Canzoniero, L.M.; Yu, S.P.; Ying, H.S.; Koh, J.Y.; Kerchner, G.A.; Choi, D.W. Measurement of intracellular free zinc in living cortical neurons: Routes of entry. J. Neurosci. 1997, 17, 9554–9564. [Google Scholar] [CrossRef] [Green Version]
- Ohana, E.; Segal, D.; Palty, R.; Ton-That, D.; Moran, A.; Sensi, S.L.; Weiss, J.H.; Hershfinkel, M.; Sekler, I. A sodium zinc exchange mechanism is mediating extrusion of zinc in mammalian cells. J. Biol. Chem. 2004, 279, 4278–4284. [Google Scholar] [CrossRef] [Green Version]
- Sekler, I.; Sensi, S.L.; Hershfinkel, M.; Silverman, W.F. Mechanism and regulation of cellular zinc transport. Mol. Med. 2007, 13, 337–343. [Google Scholar] [CrossRef]
- Lee, B.K.; Jung, Y.S. The Na+/H+ exchanger-1 inhibitor cariporide prevents glutamate-induced necrotic neuronal death by inhibiting mitochondrial Ca2+ overload. J. Neurosci Res. 2012, 90, 860–869. [Google Scholar] [CrossRef]
- Masereel, B.; Pochet, L.; Laeckmann, D. An overview of inhibitors of Na+/H+ exchanger. Eur. J. Med. Chem. 2003, 38, 547–554. [Google Scholar] [CrossRef]
- Durham-Lee, J.C.; Mokkapati, V.U.; Johnson, K.M.; Nesic, O. Amiloride improves locomotor recovery after spinal cord injury. J. Neurotrauma 2011, 28, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.G.; Zhu, X.M.; Chu, X.P.; Minami, M.; Hey, J.; Wei, W.L.; MacDonald, J.F.; Wemmie, J.A.; Price, M.P.; Welsh, M.J.; et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 2004, 118, 687–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, R.L.; Sung, M.L.; Vasylyev, D.; Zhang, M.Y.; Albinson, K.; Kubek, K.; Kagan, N.; Beyer, C.; Lin, Q.; Dwyer, J.M.; et al. Amiloride is neuroprotective in an MPTP model of Parkinson’s disease. Neurobiol. Dis. 2008, 31, 334–341. [Google Scholar] [CrossRef]
- Lazarewicz, J.W.; Rybkowski, W.; Sadowski, M.; Ziembowicz, A.; Alaraj, M.; Wegiel, J.; Wisniewski, H.M. N-methyl-D-aspartate receptor-mediated, calcium-induced calcium release in rat dentate gyrus/CA4 in vivo. J. Neurosci. Res. 1998, 51, 76–84. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Chanana, V.; Watters, J.J.; Ferrazzano, P.; Sun, D. Role of sodium/hydrogen exchanger isoform 1 in microglial activation and proinflammatory responses in ischemic brains. J. Neurochem. 2011, 119, 124–135. [Google Scholar] [CrossRef]
- Hua, J.S.; Li, L.P.; Zhu, X.M. Effects of moxibustion pretreating on SOD and MDA in the rat of global brain ischemia. J. Tradit. Chin. Med. 2008, 28, 289–292. [Google Scholar] [CrossRef]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.Y.; Jung, J.W.; Suh, S.W. The emerging role of zinc in the pathogenesis of multiple sclerosis. Int. J. Mol. Sci. 2017, 18, 2070. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Vana, A.C.; Ribeiro, R.; Zhang, Y. Distinct role of nitric oxide and peroxynitrite in mediating oligodendrocyte toxicity in culture and in experimental autoimmune encephalomyelitis. Neuroscience 2011, 184, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, H.; Li, J.; Dong, L.; Xu, P.; Chen, W.; Neve, R.L.; Volpe, J.J.; Rosenberg, P.A. Intracellular zinc release and ERK phosphorylation are required upstream of 12-lipoxygenase activation in peroxynitrite toxicity to mature rat oligodendrocytes. J. Biol. Chem. 2006, 281, 9460–9470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Dhammu, T.S.; Sakakima, H.; Shunmugavel, A.; Gilg, A.G.; Singh, A.K.; Singh, I. The inhibitory effect of S-nitrosoglutathione on blood-brain barrier disruption and peroxynitrite formation in a rat model of experimental stroke. J. Neurochem. 2012, 123, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Torreilles, F.; Salman-Tabcheh, S.; Guerin, M.; Torreilles, J. Neurodegenerative disorders: The role of peroxynitrite. Brain Res. Brain Res. Rev. 1999, 30, 153–163. [Google Scholar] [CrossRef]
- Choi, B.Y.; Lee, S.H.; Choi, H.C.; Lee, S.K.; Yoon, H.S.; Park, J.B.; Chung, W.S.; Suh, S.W. Alcohol dependence treating agent, acamprosate, prevents traumatic brain injury-induced neuron death through vesicular zinc depletion. Transl. Res. 2019, 207, 1–18. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Dalkara, T.; Gursoy-Ozdemir, Y.; Yemisci, M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011, 122, 1–9. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Thundyil, J.; Tang, S.C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef]
- Jang, B.G.; Won, S.J.; Kim, J.H.; Choi, B.Y.; Lee, M.W.; Sohn, M.; Song, H.K.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and enhances cortical neuronal injury after transient cerebral ischemia in mice. J. Trace Elem. Med. Biol. 2012, 26, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.W.; Aoyama, K.; Chen, Y.; Garnier, P.; Matsumori, Y.; Gum, E.; Liu, J.; Swanson, R.A. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J. Neurosci. 2003, 23, 10681–10690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederickson, C.J.; Kasarskis, E.J.; Ringo, D.; Frederickson, R.E. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 1987, 20, 91–103. [Google Scholar] [CrossRef]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Invest. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Kim, J.H.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Sohn, M.; Suh, S.W. Prevention of hypoglycemia-induced hippocampal neuronal death by N-acetyl-L-cysteine (NAC). Amino Acids 2017, 49, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.Y.; Lee, S.H.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of protocatechuic acid reduces traumatic brain injury-induced neuronal death. Int. J. Mol. Sci. 2017, 18, 2510. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, T.M.; Swanson, R.A. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J. Immunol. 2005, 174, 2288–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruth, R.E.; Feinerman, G.S. Foreign and endogenous serum protein extravasation during harmaline tremors or kainic acid seizures in the rat: A comparison. Acta Neuropathol. 1988, 76, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, B.S.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, D.H.; Park, M.K.; Suh, S.W. An Inhibitor of the Sodium–Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia. Int. J. Mol. Sci. 2020, 21, 4232. https://doi.org/10.3390/ijms21124232
Kang BS, Choi BY, Kho AR, Lee SH, Hong DK, Jeong JH, Kang DH, Park MK, Suh SW. An Inhibitor of the Sodium–Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia. International Journal of Molecular Sciences. 2020; 21(12):4232. https://doi.org/10.3390/ijms21124232
Chicago/Turabian StyleKang, Beom Seok, Bo Young Choi, A Ra Kho, Song Hee Lee, Dae Ki Hong, Jeong Hyun Jeong, Dong Hyeon Kang, Min Kyu Park, and Sang Won Suh. 2020. "An Inhibitor of the Sodium–Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia" International Journal of Molecular Sciences 21, no. 12: 4232. https://doi.org/10.3390/ijms21124232
APA StyleKang, B. S., Choi, B. Y., Kho, A. R., Lee, S. H., Hong, D. K., Jeong, J. H., Kang, D. H., Park, M. K., & Suh, S. W. (2020). An Inhibitor of the Sodium–Hydrogen Exchanger-1 (NHE-1), Amiloride, Reduced Zinc Accumulation and Hippocampal Neuronal Death after Ischemia. International Journal of Molecular Sciences, 21(12), 4232. https://doi.org/10.3390/ijms21124232