An Integrative Synthetic Biology Approach to Interrogating Cellular Ubiquitin and Ufm Signaling

 ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
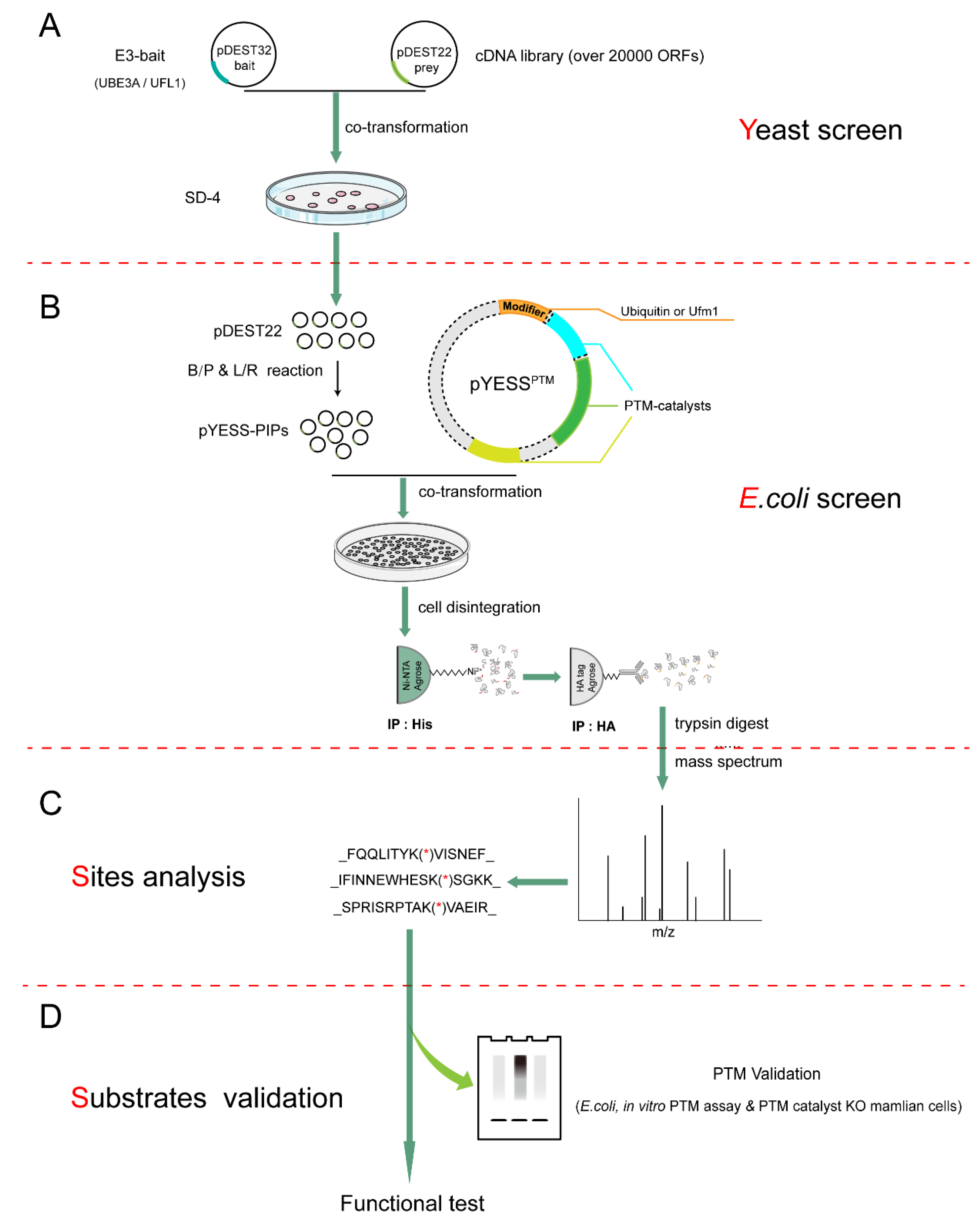
2.1. The Workflow of YESS Approach
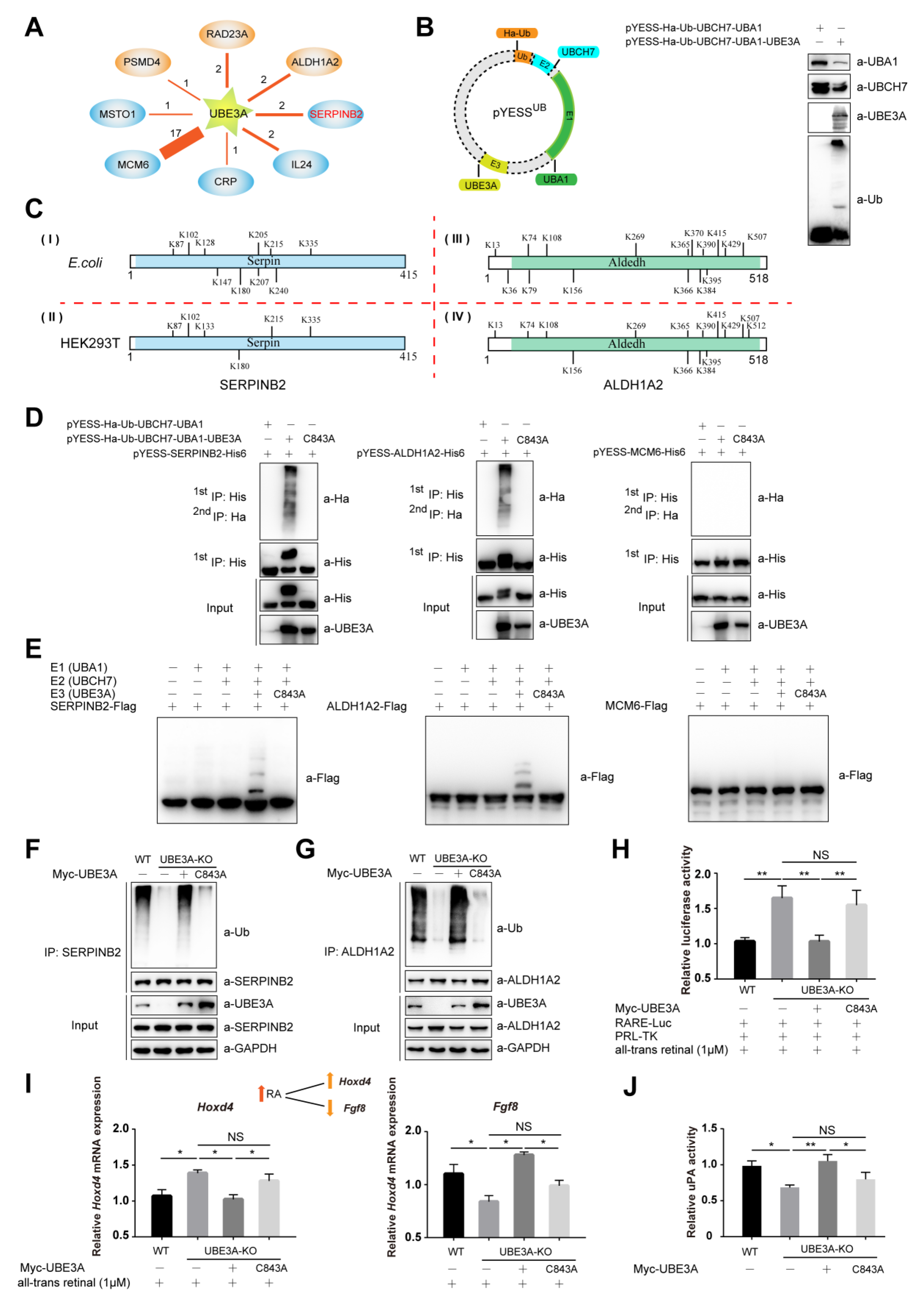
2.2. YESSUB, YESS Applied to Interrogate UBE3A-Mediated Ub Signaling
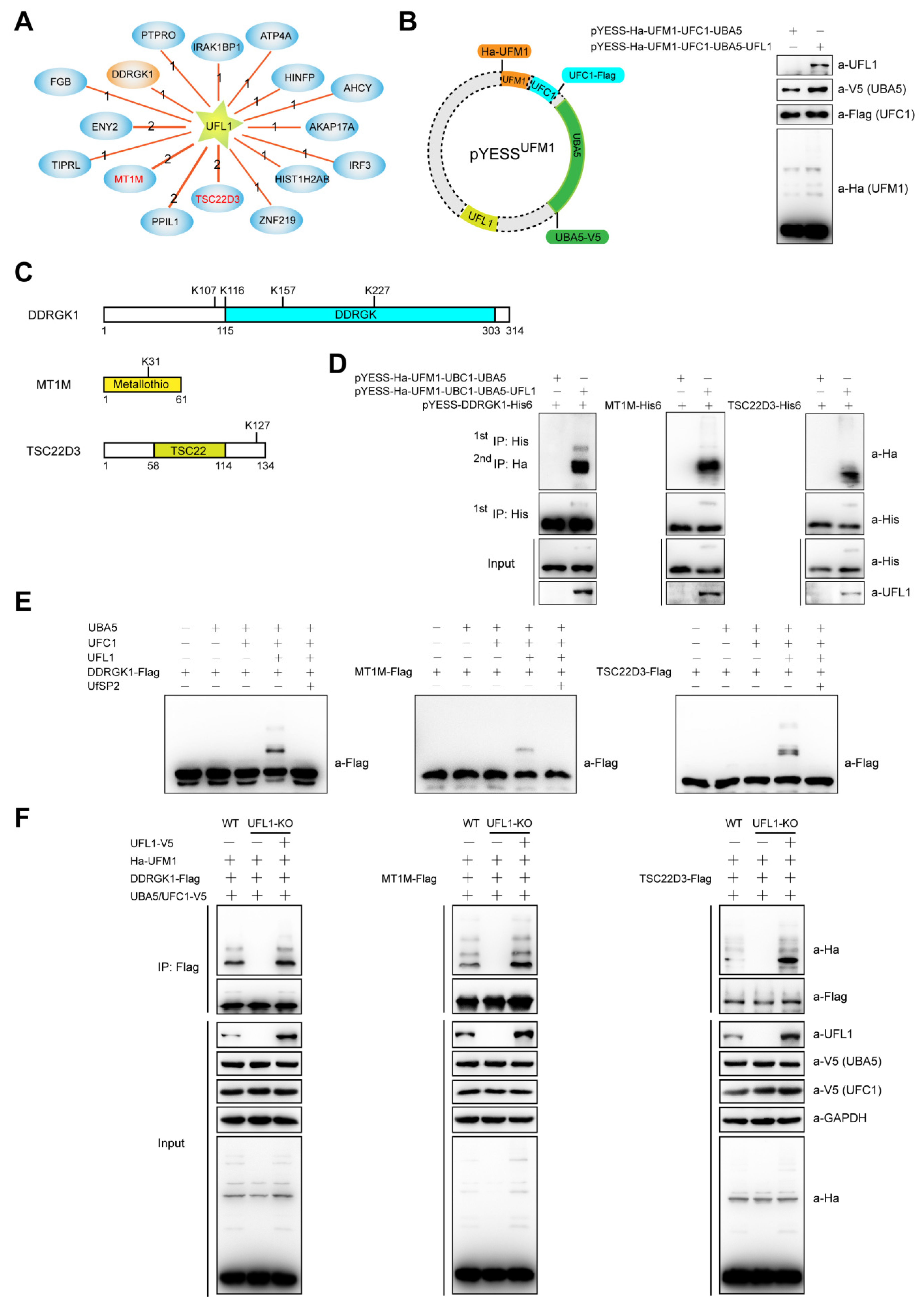
2.3. YESSUFM1, YESS Applied to Identify and Characterize Substrates for UFL1-Mediated Ufmylation
3. Discussion
4. Materials and Methods
4.1. Plasmids Construction
4.2. Yeast Two-Hybrid (Y2H) Screening
4.3. Co-Transformation and Two-Step Enrichment of the PTM Substrates in E. coli
4.4. Ubiquitination/Ufmylation Assay in Reconstituted E. coli System
4.5. Expression and Purification of Recombinant Proteins
4.6. In Vitro Ubiquitination/Ufmylation Assay
4.7. In Vivo Ubiquitination/Ufmylation Assay
4.8. Two-Step Enrichments of the Proteins from Mammalian Cells
4.9. Mass Spectrometry Analysis to Map the Ubiquitination/Ufmylation Sites on Substrates
4.10. Cell Culture and Transfection
4.11. Generation of UBE3A/UFL1-Ablated Cell Lines
- UBE3A forward primers: 5′-CACCGagcacaaaactcattcgtgc-3′,
- UBE3A reverse primers: 5′-AAACgcacgaatgagttttgtgctC-3′.
- UFL1 forward primers: 5′-CACCG ccagcgggcgcagttcgccg-3′,
- UFL1 reverse primers: 5′-AAAC cggcgaactgcgcccgctggC-3′.
4.12. Co-Immunoprecipitation and Immunoblotting Assay
4.13. Immunofluorescence Microscopy
4.14. GST Pull-Down Assay
4.15. Luciferase Assay
4.16. Quantitative Real-Time PCR (qPCR)
4.17. uPA Activity Assay
4.18. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A.; Varshavsky, A. Basic Medical Research Award. The ubiquitin system. Nat. Med. 2000, 6, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Hochstrasser, M. Recent progress in ubiquitin and ubiquitin-like protein (Ubl) signaling. Cell Res. 2016, 26, 389–390. [Google Scholar] [CrossRef] [Green Version]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.H.; Kim, H.Y.; Heo, A.J.; Lee, S.H.; Lee, M.J.; Kim, S.B.; Srinivasrao, G.; Mun, S.R.; Cha-Molstad, H.; Ciechanover, A.; et al. The N-Degron Pathway Mediates ER-phagy. Mol. Cell 2019, 75, 1058–1072. [Google Scholar] [CrossRef]
- Chou, T.F.; Brown, S.J.; Minond, D.; Nordin, B.E.; Li, K.; Jones, A.C.; Chase, P.; Porubsky, P.R.; Stoltz, B.M.; Schoenen, F.J.; et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 4834–4839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swatek, K.N.; Usher, J.L.; Kueck, A.F.; Gladkova, C.; Mevissen, T.E.T.; Pruneda, J.N.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Christensen, D.E.; Brzovic, P.S.; Klevit, R.E. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol. 2007, 14, 941–948. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Love, I.M.; Grossman, S.R. It Takes 15 to Tango: Making Sense of the Many Ubiquitin Ligases of p53. Genes Cancer 2012, 3, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, P.; Shao, X.; Yang, Y.; Liu, X.; Feng, M.; Yu, Q.; Hu, R.; Wang, Z. The E3 Ligase RING1 Targets p53 for Degradation and Promotes Cancer Cell Proliferation and Survival. Cancer Res. 2018, 78, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, K.; Sou, Y.S.; Tada, N.; Nakamura, E.; Iemura, S.; Natsume, T.; Kang, S.H.; Chung, C.H.; Kasahara, M.; Kominami, E.; et al. A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem. 2010, 285, 5417–5427. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, Y.; Song, L.; Zeng, L.; Yi, W.; Liu, T.; Chen, H.; Wang, M.; Ju, Z.; Cong, Y.S. A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1alpha stability. Nat. Commun. 2017, 8, 14186. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, Y.; Rogers, H.; Saidi, L.; Noguchi, C.T.; Li, H.; Yewdell, J.W.; Guydosh, N.R.; Ye, Y. UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 2019, 30, 5–20. [Google Scholar] [CrossRef]
- Qin, B.; Yu, J.; Nowsheen, S.; Wang, M.; Tu, X.; Liu, T.; Li, H.; Wang, L.; Lou, Z. UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Walczak, C.P.; Leto, D.E.; Zhang, L.; Riepe, C.; Muller, R.Y.; DaRosa, P.A.; Ingolia, N.T.; Elias, J.E.; Kopito, R.R. Ribosomal protein RPL26 is the principal target of UFMylation. Proc. Natl. Acad. Sci. USA 2019, 116, 1299–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.M.; Kang, S.H.; Kim, J.Y.; Lee, J.E.; Seong, M.W.; Lee, S.W.; Ka, S.H.; Sou, Y.S.; Komatsu, M.; Tanaka, K.; et al. Modification of ASC1 by UFM1 is crucial for ERalpha transactivation and breast cancer development. Mol. Cell 2014, 56, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iconomou, M.; Saunders, D.N. Systematic approaches to identify E3 ligase substrates. Biochem. J. 2016, 473, 4083–4101. [Google Scholar] [CrossRef] [Green Version]
- Keren-Kaplan, T.; Attali, I.; Motamedchaboki, K.; Davis, B.A.; Tanner, N.; Reshef, Y.; Laudon, E.; Kolot, M.; Levin-Kravets, O.; Kleifeld, O.; et al. Synthetic biology approach to reconstituting the ubiquitylation cascade in bacteria. EMBO J. 2012, 31, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin-Kravets, O.; Tanner, N.; Shohat, N.; Attali, I.; Keren-Kaplan, T.; Shusterman, A.; Artzi, S.; Varvak, A.; Reshef, Y.; Shi, X.; et al. A bacterial genetic selection system for ubiquitylation cascade discovery. Nat. Methods 2016, 13, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.M.; Isasa, M.; Ordureau, A.; Prado, M.A.; Beausoleil, S.A.; Jedrychowski, M.P.; Finley, D.J.; Harper, J.W.; Gygi, S.P. Highly Multiplexed Quantitative Mass Spectrometry Analysis of Ubiquitylomes. Cell Syst. 2016, 3, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.P.; Yates, J.R., III. Recent trends of capillary electrophoresis-mass spectrometry in proteomics research. Mass Spectrom. Rev. 2019, 38, 445–460. [Google Scholar] [CrossRef]
- Mann, M. The ever expanding scope of electrospray mass spectrometry—A 30 year journey. Nat. Commun. 2019, 10, 3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Sell, G.L.; Margolis, S.S. From UBE3A to Angelman syndrome: A substrate perspective. Front. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, S.J.; Laufer, B.I.; Beitnere, U.; Berg, E.L.; Silverman, J.L.; O’Geen, H.; Segal, D.J.; LaSalle, J.M. Imprinting effects of UBE3A loss on synaptic gene networks and Wnt signaling pathways. Hum. Mol. Genet. 2019, 28, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Reynolds, K.; Ji, Y.; Gu, R.; Rai, S.; Zhou, C.J. Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. J. Neurodev. Disord. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Furumai, R.; Tamada, K.; Liu, X.; Takumi, T. UBE3A regulates the transcription of IRF, an antiviral immunity. Hum. Mol Genet. 2019, 28, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.J.; Segal, D.J.; LaSalle, J.M. UBE3A: An E3 Ubiquitin Ligase with Genome-Wide Impact in Neurodevelopmental Disease. Front. Mol. Neurosci. 2018, 11, 476. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, P.; Gao, H.; Gu, Y.; Yang, J.; Peng, H.; Xu, X.; Wang, H.; Yang, M.; Liu, X.; et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell 2014, 26, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Peng, J. Characterization of polyubiquitin chain structure by middle-down mass spectrometry. Anal. Chem. 2008, 80, 3438–3444. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, A.D.; MacFadden, A.; Wu, Z.; Peng, J.; Liu, C.W. Autoregulation of the 26S proteasome by in situ ubiquitination. Mol. Biol. Cell 2014, 25, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Talis, A.L.; Howley, P.M. Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 1999, 274, 18785–18792. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 169, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Li, C.; Gao, X.; Xia, K.; Guo, H.; Li, Y.; Hao, Z.; Zhang, L.; Gao, D.; Xu, C.; et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2018, 28, 48–68. [Google Scholar] [CrossRef]
- Yu, H.; Maurer, F.; Medcalf, R.L. Plasminogen activator inhibitor type 2: A regulator of monocyte proliferation and differentiation. Blood 2002, 99, 2810–2818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.; Yu, H.; Wang, C.; Sun, L.; Jiang, W.; Zhang, P.; Xiao, Q.; Han, D.; Saiyin, H.; Zhu, J.; et al. Metallothionein MT1M is a tumor suppressor of human hepatocellular carcinomas. Carcinogenesis 2012, 33, 2568–2577. [Google Scholar] [CrossRef] [Green Version]
- DeJesus, R.; Moretti, F.; McAllister, G.; Wang, Z.; Bergman, P.; Liu, S.; Frias, E.; Alford, J.; Reece-Hoyes, J.S.; Lindeman, A.; et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. eLife 2016, 5, e17290. [Google Scholar] [CrossRef] [PubMed]
- Nahorski, M.S.; Maddirevula, S.; Ishimura, R.; Alsahli, S.; Brady, A.F.; Begemann, A.; Mizushima, T.; Guzman-Vega, F.J.; Obata, M.; Ichimura, Y.; et al. Biallelic UFM1 and UFC1 mutations expand the essential role of ufmylation in brain development. Brain J. Neurol. 2018, 141, 1934–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burande, C.F.; Heuze, M.L.; Lamsoul, I.; Monsarrat, B.; Uttenweiler-Joseph, S.; Lutz, P.G. A label-free quantitative proteomics strategy to identify E3 ubiquitin ligase substrates targeted to proteasome degradation. Mol. Cell. Proteom. MCP 2009, 8, 1719–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, K.G.; Simonetta, M.; Maiolica, A.; Seller, C.A.; Toczyski, D.P. Ubiquitin ligase trapping identifies an SCF(Saf1) pathway targeting unprocessed vacuolar/lysosomal proteins. Mol. Cell 2014, 53, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Bhuripanyo, K.; Wang, Y.; Liu, X.; Zhou, L.; Liu, R.; Duong, D.; Zhao, B.; Bi, Y.; Zhou, H.; Chen, G.; et al. Identifying the substrate proteins of U-box E3s E4B and CHIP by orthogonal ubiquitin transfer. Sci. Adv. 2018, 4, e1701393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumimoto, K.; Nakayama, K.I. Recent insight into the role of FBXW7 as a tumor suppressor. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Deol, K.K.; Lorenz, S.; Strieter, E.R. Enzymatic Logic of Ubiquitin Chain Assembly. Front. Physiol. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.K.; Kolch, W.; Kholodenko, B.N. When ubiquitination meets phosphorylation: A systems biology perspective of EGFR/MAPK signalling. Cell Commun. Signal. CCS 2013, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Yang, J.; Li, G.; You, Q.; Han, W.; Li, T.; Gao, D.; Xie, X.; Lee, B.H.; Du, J.; et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017, 27, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Han, T.; Guo, R.; Chen, P.; Peng, C.; Prag, G.; Hu, R. An Integrative Synthetic Biology Approach to Interrogating Cellular Ubiquitin and Ufm Signaling. Int. J. Mol. Sci. 2020, 21, 4231. https://doi.org/10.3390/ijms21124231
Li C, Han T, Guo R, Chen P, Peng C, Prag G, Hu R. An Integrative Synthetic Biology Approach to Interrogating Cellular Ubiquitin and Ufm Signaling. International Journal of Molecular Sciences. 2020; 21(12):4231. https://doi.org/10.3390/ijms21124231
Chicago/Turabian StyleLi, Chuanyin, Tianting Han, Rong Guo, Peng Chen, Chao Peng, Gali Prag, and Ronggui Hu. 2020. "An Integrative Synthetic Biology Approach to Interrogating Cellular Ubiquitin and Ufm Signaling" International Journal of Molecular Sciences 21, no. 12: 4231. https://doi.org/10.3390/ijms21124231
APA StyleLi, C., Han, T., Guo, R., Chen, P., Peng, C., Prag, G., & Hu, R. (2020). An Integrative Synthetic Biology Approach to Interrogating Cellular Ubiquitin and Ufm Signaling. International Journal of Molecular Sciences, 21(12), 4231. https://doi.org/10.3390/ijms21124231