CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
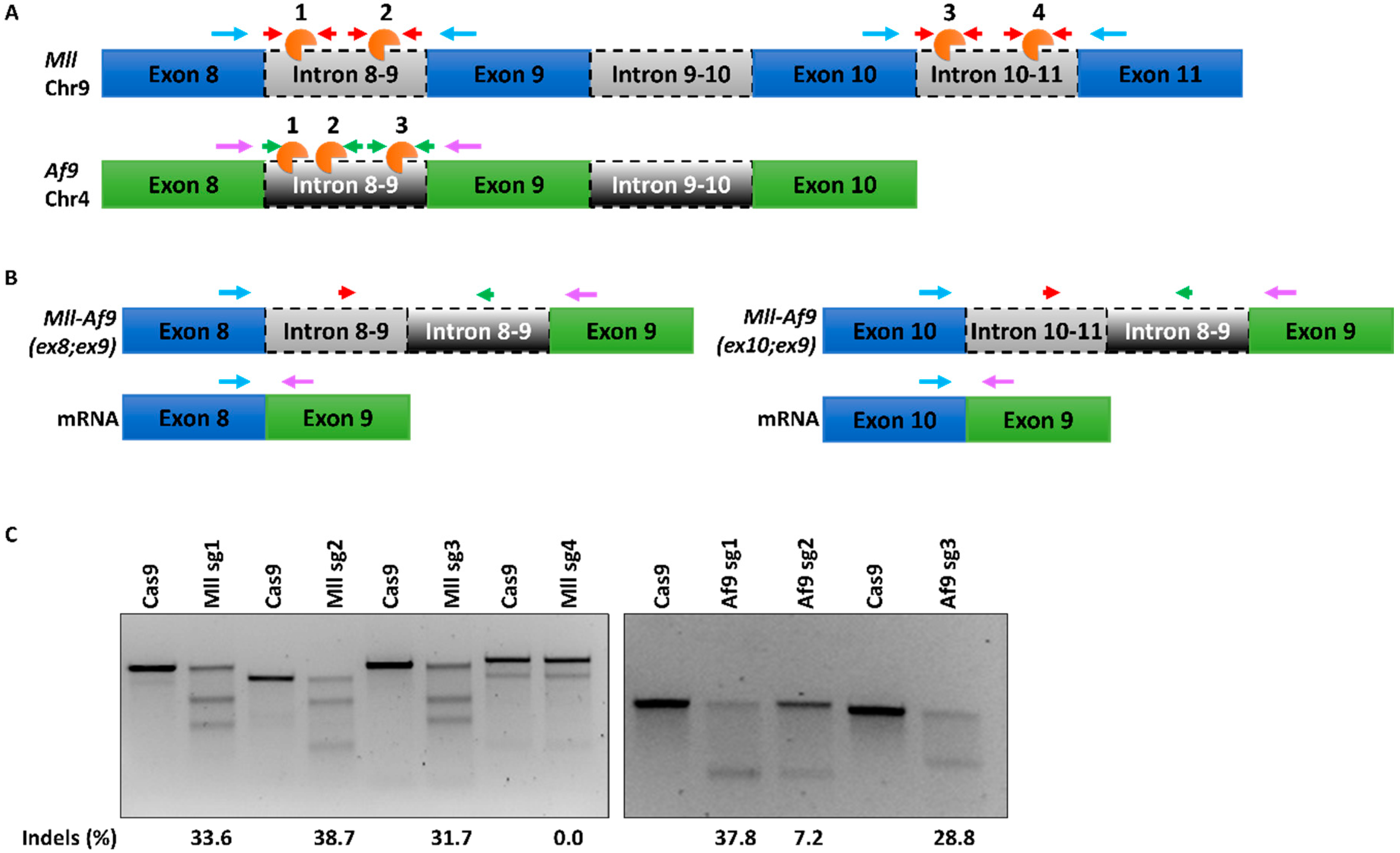
2.1. A CRISPR/Cas9 System for Mll-Af9 Genome Editing (302)
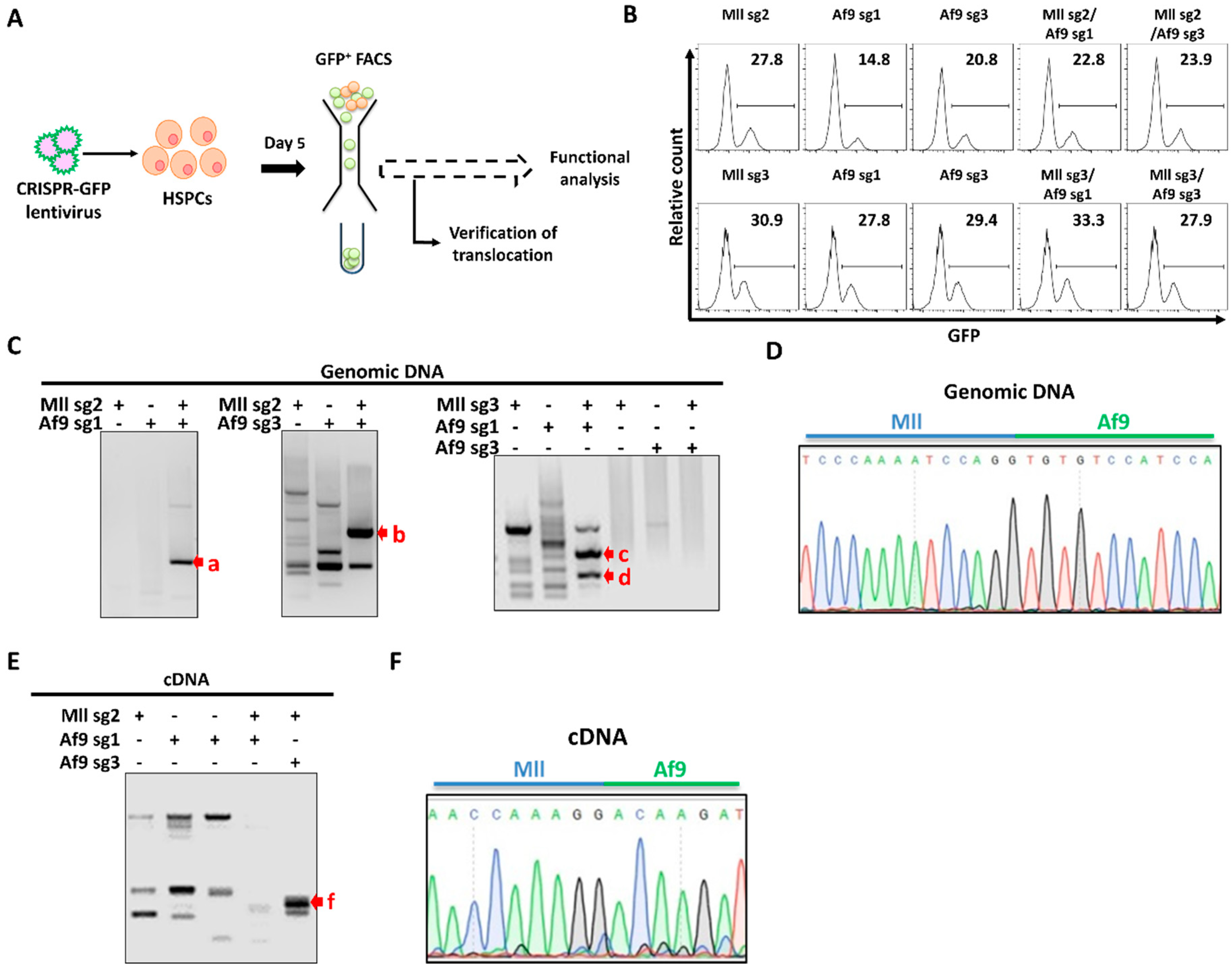
2.2. Endogenous Generation of MA9 Translocation in Murine HSPCs (368)
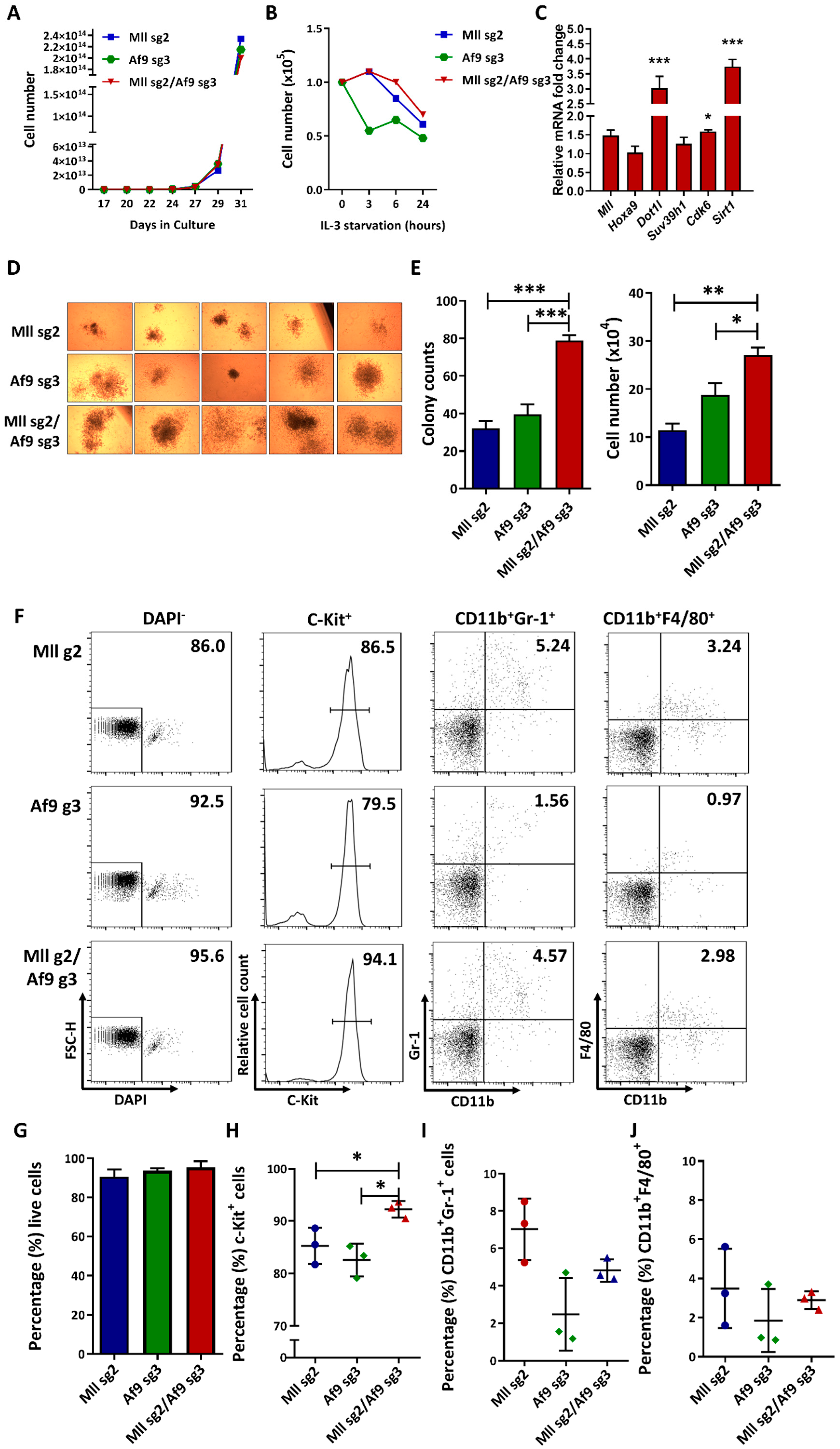
2.3. CRISPR-Mediated MA9 Translocations Recapitulate Features of MA9 Leukaemia In Vitro (622)
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Murine Bone Marrow (BM) Harvest and c-Kit Enrichment
4.3. sgRNA Design, Synthesis and CRISPR Transduction
4.4. Flow Cytometry and Cell Sorting
4.5. Surveyor Assay
4.6. Genomic Target Amplification (GTA)
4.7. Reverse Transcription PCR
4.8. Gene Expression Analysis (qRT-PCR)
4.9. Colony-Forming Cell (CFC) Assay
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slany, R.K. The molecular mechanics of mixed lineage leukemia. Oncogene 2016, 35, 5215–5223. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Jasin, M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 2000, 405, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Slany, R.K. When epigenetics kills: MLL fusion proteins in leukemia. Hematol. Oncol. 2005, 23, 1–9. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; De Braekeleer, E.; De Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef]
- Huret, J.L.; Ahmad, M.; Arsaban, M.; Bernheim, A.; Cigna, J.; Desangles, F.; Guignard, J.C.; Jacquemot-Perbal, M.C.; Labarussias, M.; Leberre, V.; et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013, 41, D920–D924. [Google Scholar] [CrossRef]
- Super, H.J.G.; McCabe, N.R.; Thirman, M.J.; Larson, R.A.; Le Beau, M.M.; Pedersen- Bjergaard, J.; Philip, P.; Diaz, M.O.; Rowley, J.D. Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 1993, 82, 3705–3711. [Google Scholar] [CrossRef] [Green Version]
- Malik, B.; Hemenway, C.S. CBX8, a component of the Polycomb PRC1 complex, modulates DOT1L-mediated gene expression through AF9/MLLT3. FEBS Lett. 2013, 587, 3038–3044. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xia, X.; Reisenauer, M.R.; Hemenway, C.S.; Kone, B.C. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J. Biol. Chem. 2006, 281, 18059–18068. [Google Scholar] [CrossRef] [Green Version]
- Erfurth, F.; Hemenway, C.S.; de Erkenez, A.C.; Domer, P.H. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia 2004, 18, 92–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, B.I.; Kuntimaddi, A.; Schmidt, C.R.; Cierpicki, T.; Johnson, S.A.; Bushweller, J.H. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure 2013, 21, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; García-Cuéllar, M.P.; Bach, C.; Buhl, S.; Maethner, E.; Slany, R.K. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009, 7, e1000249. [Google Scholar] [CrossRef] [Green Version]
- Kuntimaddi, A.; Achille, N.J.; Thorpe, J.; Lokken, A.A.; Singh, R.; Hemenway, C.S.; Adli, M.; Zeleznik-Le, N.J.; Bushweller, J.H. Degree of Recruitment of DOT1L to MLL-AF9 Defines Level of H3K79 Di- and Tri-methylation on Target Genes and Transformation Potential. Cell Rep. 2015, 11, 808–820. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9—Mediated leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Somervaille, T.C.P.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; Den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Zeisig, B.B.; Milne, T.; Garcia-Cuellar, M.-P.; Schreiner, S.; Martin, M.-E.; Fuchs, U.; Borkhardt, A.; Chanda, S.K.; Walker, J.; Soden, R.; et al. Hoxa9 and Meis1 Are Key Targets for MLL-ENL-Mediated Cellular Immortalization. Mol. Cell. Biol. 2004, 24, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Chu, Y.; Wang, L.; Chen, X.; Chen, Y.; Cheng, H.; Zhang, L.; Zhou, Y.; Yang, F.C.; Cheng, T.; et al. PBX3 is essential for leukemia stem cell maintenance in MLL-rearranged leukemia. Int. J. Cancer 2017, 141, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Prange, K.H.M.; Mandoli, A.; Kuznetsova, T.; Wang, S.Y.; Sotoca, A.M.; Marneth, A.E.; Van Der Reijden, B.A.; Stunnenberg, H.G.; Martens, J.H.A. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene 2017, 36, 3346–3356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.R.; Hudson, W.A.; Chen, W.; Nishiuchi, R.; Yao, Q.; Kersey, J.H. Hoxa9 influences the phenotype but not the incidence of Mll-AF9 fusion gene leukemia. Blood 2004, 103, 1823–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Linden, M.H.; Willekes, M.; Roon, E.; Seslija, L.; Schneider, P.; Pieters, R.; Stam, R.W. MLL fusion-driven activation of CDK6 potentiates proliferation in MLL-rearranged infant ALL. Cell Cycle 2014, 13, 834–844. [Google Scholar] [CrossRef] [Green Version]
- Bindels, E.M.J.; Havermans, M.; Lugthart, S.; Erpelinck, C.; Wocjtowicz, E.; Krivtsov, A.V.; Rombouts, E.; Armstrong, S.A.; Taskesen, E.; Haanstra, J.R.; et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood 2012, 119, 5838–5849. [Google Scholar] [CrossRef] [PubMed]
- Laricchia-Robbio, L.; Nucifora, G. Significant increase of self-renewal in hematopoietic cells after forced expression of EVI1. Blood Cells Mol. Dis. 2008, 40, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.F.; Wu, G.; Mi, S.; He, F.; Wu, J.; Dong, J.; Luo, R.T.; Mattison, R.; Kaberlein, J.J.; Prabhakar, S.; et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 2011, 117, 6895–6905. [Google Scholar] [CrossRef] [Green Version]
- Takacova, S.; Luzna, P.; Stranecky, V.; Divoky, V. The Potential Role of the Six1/Eya1 Pathway in the Establishment of Leukemia Stem Cells in MLL-ENL—Induced Leukemia. Blood 2011, 118, 1362. [Google Scholar] [CrossRef]
- Arai, S.; Yoshimi, A.; Shimabe, M.; Ichikawa, M.; Nakagawa, M.; Imai, Y.; Goyama, S.; Kurokawa, M. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood 2011, 117, 6304–6314. [Google Scholar] [CrossRef] [Green Version]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.V.; et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood 2014, 124, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.J.; King, G.; Rabbitts, T.H. An MII-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Dobson, C.L.; Warren, A.J.; Pannell, R.; Forster, A.; Lavenir, I.; Corral, J.; Smith, A.J.H.; Rabbitts, T.H. The Mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 1999, 18, 3564–3574. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Q.; Hudson, W.A.; Kumar, A.; Kirchhof, N.; Kersey, J.H. Amurine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood 2006, 108, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Kumar, A.R.; Hudson, W.A.; Li, Q.; Wu, B.; Staggs, R.A.; Lund, E.A.; Sam, T.N.; Kersey, J.H. Malignant Transformation Initiated by Mll-AF9: Gene Dosage and Critical Target Cells. Cancer Cell 2008, 13, 432–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, E.C.; Pannell, R.; Simpson, E.M.; Forster, A.; Rabbitts, T.H. Inter-chromosomal recombination of Mll and Af9 genes mediated by cre-lox in mouse development. EMBO Rep. 2000, 1, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Drynan, L.F.; Pannell, R.; Forster, A.; Chan, N.M.M.; Cano, F.; Daser, A.; Rabbitts, T.H. Mll fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis. EMBO J. 2005, 24, 3136–3146. [Google Scholar] [CrossRef] [Green Version]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Somervaille, T.C.P.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Figueroa, M.E.; Sinha, A.U.; Stubbs, M.C.; Feng, Z.; Valk, P.J.M.; Delwel, R.; Döhner, K.; Bullinger, L.; Kung, A.L.; et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 2013, 27, 852–860. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Burkhardt, D.B.; Hartman, A.A.; Hu, X.; Eastman, A.E.; Sun, C.; Wang, X.; Zhong, M.; Krishnaswamy, S.; Guo, S. MLL-AF9 initiates transformation from fast-proliferating myeloid progenitors. Nat. Commun. 2019, 10, 5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buechele, C.; Breese, E.H.; Schneidawind, D.; Lin, C.H.; Jeong, J.; Duque-Afonso, J.; Wong, S.H.K.; Smith, K.S.; Negrin, R.S.; Porteus, M.; et al. MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood 2015, 126, 1683–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breese, E.H.; Buechele, C.; Dawson, C.; Cleary, M.L.; Porteus, M.H. Use of genome engineering to create patient specific MLL translocations in primary human hematopoietic stem and progenitor cells. PLoS ONE 2015, 10, e0136644. [Google Scholar] [CrossRef] [PubMed]
- Adikusuma, F.; Williams, N.; Grutzner, F.; Hughes, J.; Thomas, P. Targeted Deletion of an Entire Chromosome Using CRISPR/Cas9. Mol. Ther. 2017, 25, 1736–1738. [Google Scholar] [CrossRef] [PubMed]
- Maddalo, D.; Manchado, E.; Concepcion, C.P.; Bonetti, C.; Vidigal, J.A.; Han, Y.C.; Ogrodowski, P.; Crippa, A.; Rekhtman, N.; De Stanchina, E.; et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 2014, 516, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Martin, M.C.; Garcia, A.; Cigudosa, J.C.; Ramirez, J.C.; Rodriguez-Perales, S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 2014, 5, 3964. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zhang, L.; Zhou, X.; Chen, X.; Huang, G.; Li, F.; Wang, R.; Wu, N.; Yan, Y.; Tong, C.; et al. Induction of site-specific chromosomal translocations in embryonic stem cells by CRISPR/Cas9. Sci. Rep. 2016, 6, 21918. [Google Scholar] [CrossRef]
- Reimer, J.; Knöß, S.; Labuhn, M.; Charpentier, E.M.; Göhring, G.; Schlegelberger, B.; Klusmann, J.H.; Heckl, D. CRISPR-Cas9-induced t(11;19)/MLL-ENL translocations initiate leukemia in human hematopoietic progenitor cells in vivo. Haematologica 2017, 102, 1558–1566. [Google Scholar] [CrossRef] [Green Version]
- Schneidawind, C.; Jeong, J.; Schneidawind, D.; Kim, I.S.; Duque-Afonso, J.; Wong, S.H.K.; Iwasaki, M.; Breese, E.H.; Zehnder, J.L.; Porteus, M.; et al. MLL leukemia induction by t(9;11) chromosomal translocation in human hematopoietic stem cells using genome editing. Blood Adv. 2018, 2, 832–845. [Google Scholar] [CrossRef]
- Jansen, M.W.J.C.; van der Velden, V.H.J.; van Dongen, J.J.M. Efficient and easy detection of MLL-AF4, MLL-AF9 and MLL-ENL fusion gene transcripts by multiplex real-time quantitative RT-PCR in TaqMan and LightCycler [11,12]. Leukemia 2005, 19, 2016–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.; Glaser, S.P.; Xu, Z.; Di Rago, L.; Mifsud, S. Reversible growth factor dependency and autonomy during murine myelomonocytic leukemia induced by oncogenes. Proc. Natl. Acad. Sci. USA 2013, 110, 17029–17034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasca, D.; Hähnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, H.; Kanakura, Y.; Tamaki, T.; Kuriu, A.; Kitayama, H.; Ishikawa, J.; Kanayama, Y.; Yonezawa, T.; Tarui, S.; Griffin, J.D. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood 1991, 78, 2962–2968. [Google Scholar] [CrossRef] [Green Version]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrózek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): A Cancer and Leukemia Group B study. J. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef]
- Ohlsson, E.; Hasemann, M.S.; Willer, A.; Lauridsen, F.K.B.; Rapin, N.; Jendholm, J.; Porse, B.T. Initiation of MLL-rearranged AML is dependent on C/EBPα. J. Exp. Med. 2014, 211, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.J.; Wu, H.; Achille, N.J.; Reisenauer, M.R.; Chou, C.W.; Zeleznik-Le, N.J.; Hemenway, C.S.; Zhang, W. Histone H3 Lysine 79 Methyltransferase Dot1 Is Required for Immortalization by MLL Oncogenes. Cancer Res. 2010, 70, 10234–10242. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Jager, A.; Domizi, P.; Pavel-Dinu, M.; Gojenola, L.; Iwasaki, M.; Wei, M.C.; Pan, F.; Zehnder, J.L.; Porteus, M.H.; et al. High-efficiency CRISPR induction of t(9;11) chromosomal translocations and acute leukemias in human blood stem cells. Blood Adv. 2019, 3, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Yalla, K.; Salomé, M.; Ananyambica Moka, H.; Gómez Castañeda, E.; Eyers, P.A.; Keeshan, K. Trib2 expression in granulocyte-monocyte progenitors drives a highly drug resistant acute myeloid leukaemia linked to elevated Bcl2. Oncotarget 2018, 9, 14977–14992. [Google Scholar]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarrou, E.; Richmond, L.; Carmody, R.J.; Gibson, B.; Keeshan, K. CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis. Int. J. Mol. Sci. 2020, 21, 4266. https://doi.org/10.3390/ijms21124266
Sarrou E, Richmond L, Carmody RJ, Gibson B, Keeshan K. CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis. International Journal of Molecular Sciences. 2020; 21(12):4266. https://doi.org/10.3390/ijms21124266
Chicago/Turabian StyleSarrou, Evgenia, Laura Richmond, Ruaidhrí J. Carmody, Brenda Gibson, and Karen Keeshan. 2020. "CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis" International Journal of Molecular Sciences 21, no. 12: 4266. https://doi.org/10.3390/ijms21124266
APA StyleSarrou, E., Richmond, L., Carmody, R. J., Gibson, B., & Keeshan, K. (2020). CRISPR Gene Editing of Murine Blood Stem and Progenitor Cells Induces MLL-AF9 Chromosomal Translocation and MLL-AF9 Leukaemogenesis. International Journal of Molecular Sciences, 21(12), 4266. https://doi.org/10.3390/ijms21124266