Lineage Decision-Making within Normal Haematopoietic and Leukemic Stem Cells



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
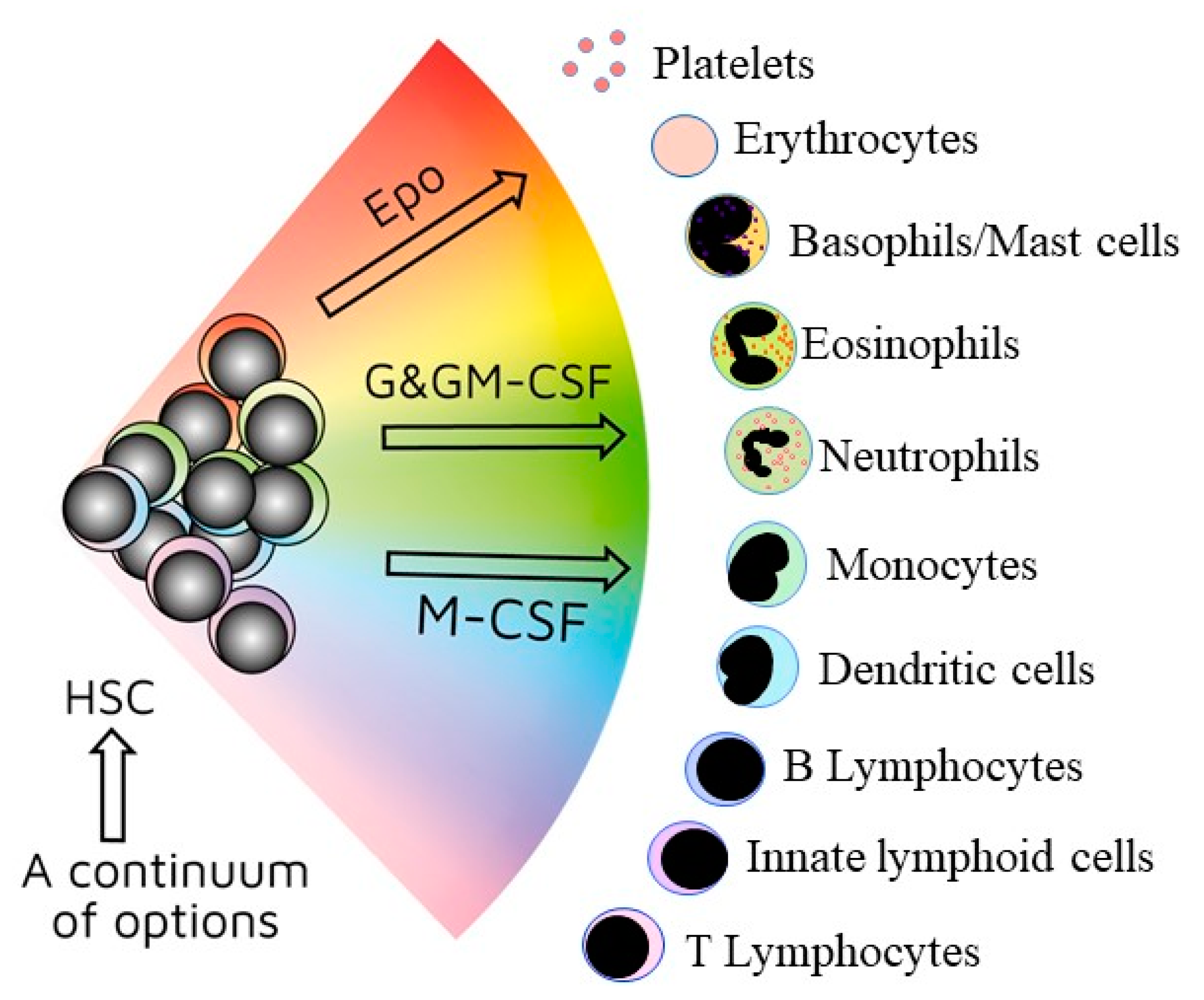
2. The Nature of Haematopoietic Stem Cells
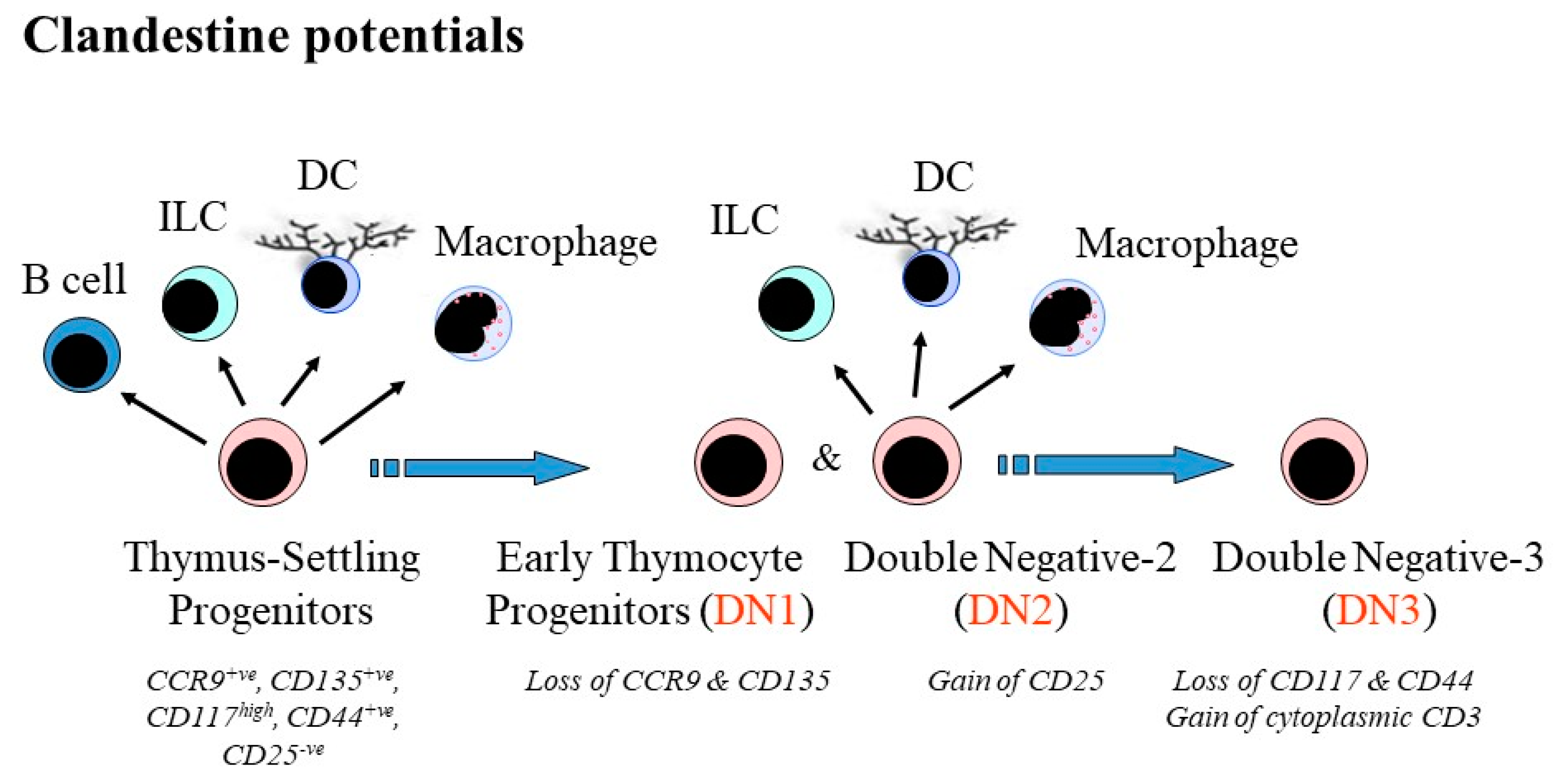
3. Crossing Barriers and “Stepping Sideways” to an Alternative Pathway
4. The Bulk of Leukaemia Cells belong to just One Cell Lineage
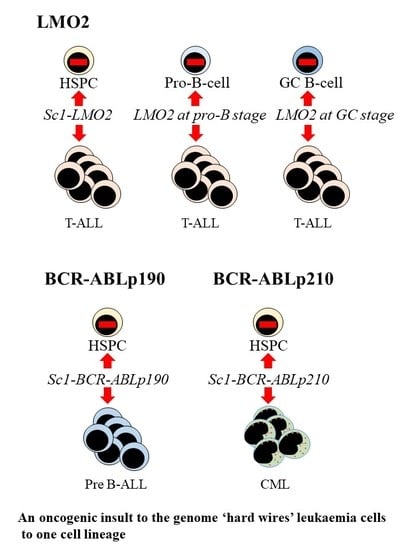
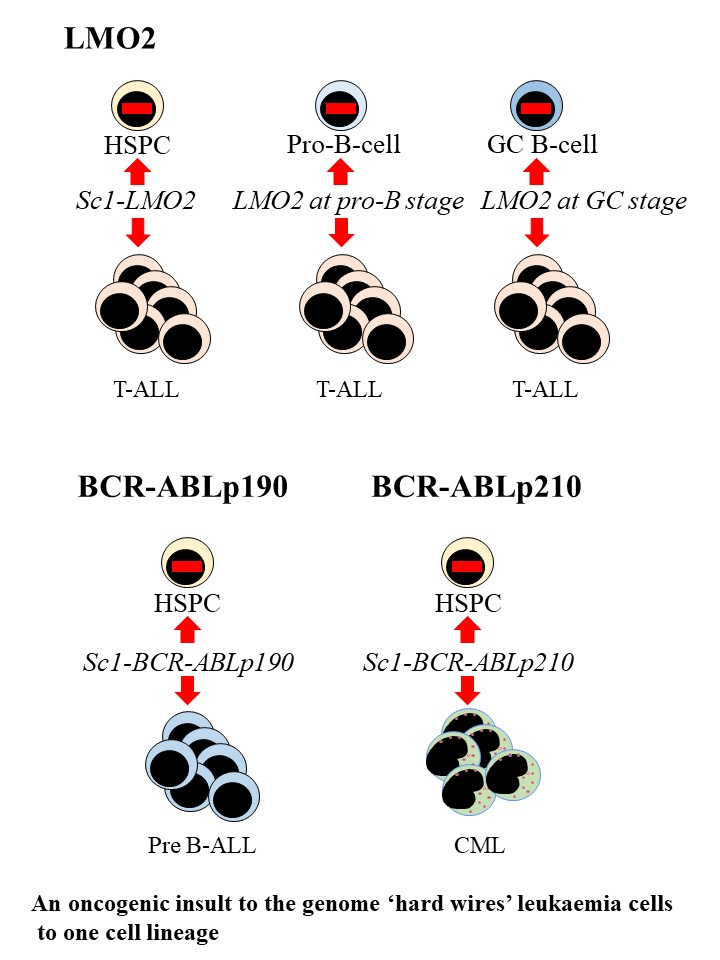
5. Oncogenic Programming of Stem and Progenitor Cells to a Single Cell Lineage
6. How Might Hard Wiring of Leukaemia Stem Cells to One Cell Lineage Occur?
7. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Halley, J.D.; Winkler, D.A.; Burden, F.R. Towards a Rosetta stone for the stem cell genome: Stochastic gene expression, network architecture, and external influences. Stem Cell Res. 2008, 1, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceredig, R.; Rolink, A.G.; Brown, G. Models of haematopoiesis: Seeing the wood for the trees. Nat. Rev. Immunol. 2009, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Kamath, M.B.; Houston, I.B.; Janovski, A.J.; Gowrisankar, S.; Jegga, A.G.; DeKoter, R.P. Dose-dependent repression of T-cell and natural killer cell genes by PU.1 enforces myeloid and B-cell identity. Leukemia 2008, 22, 1214–1225.6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontelo, P.; Manwani, D.; Galdass, M.; Karsunky, H.; Lohman, F.; Gallagher, P.G.; Bieker, J.J. Novel role for EKLF in megakaryocyte lineage commitment. Blood 2007, 110, 3871–3880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szuf, M.; McGowan, P.; Meaney, M.J. The social environment and the eigenome. Enviro. Mol. Mutagen. 2008, 49, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Arney, K.L.; Fisher, A.G. Epigenetic aspects of differentiation. J. Cell Sci. 2004, 117, 4355–4362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceredig, R. When one cell is enough. Stem Cell Res. Ther. 2012, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Doulatove, S.; Laurenti, E.; Poeppl, A.; Jurisica, I.; Dick, J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011, 333, 218–221. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanised mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Weissman, I.L.; Anderson, D.J.; Gage, F. Stem and progenitor cells: Origins, phenotypes, lineage commitments, and transdifferentiations. Annu. Rev. Cell Dev. Biol. 2001, 17, 387–403. [Google Scholar] [CrossRef] [Green Version]
- Hofer, T.; Busch, K.; Klapproth, K.; Rodewald, H.R. Fate Mapping and Quantitation of Hematopoiesis In Vivo. Annu. Rev. Immunol. 2016, 34, 449–478. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, 2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, C.J.; Cunningham, A.; Tsapogas, P.; Toellner, K.M.; Brown, G. Selective expression of FLT3 within the mouse hematopoietic stem cell compartment. Int. J. Mol. Sci. 2017, 18, 1037. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Ceredig, R. Modelling the hematopoietic landscape. Front. Cell Dev. Biol. 2019, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Carmargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, 3381. [Google Scholar] [CrossRef]
- Hu, M.; Krause, D.; Greaves, M.; Sharkis, S.; Dexter, M.; Heyworth, C.; Enver, T. Multilineage gene expression precedes commitment in the hematopoietic system. Genes Dev. 1997, 11, 774–785. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Greber, B.; Araúzo-Bravo, M.J.; Meyer, j.; Park, K.I.; Zaehres, H.; Schöler, H.R. Direct reprogramming of human neural stem cells by OCT4. Nature 2009, 461, 649–653. [Google Scholar] [CrossRef]
- Nestorowa, S.; Hamey, F.K.; Pijuan Sala, B.; Diamanti, E.; Shepherd, M.; Laurenti, E.; Wilson, N.K.; Kent, D.G.; Gottens, B. A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 2016, 128, e20–e31. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Mead, A.J. Single-cell approaches reveal novel cellular pathways for megakaryocyte and erythroid differentiation. Blood 2019, 133, 1427–1435. [Google Scholar] [CrossRef]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Buss, E.C.; Nowak, D.; Boch, T.; et al. Human hematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, R.; Morita, Y.; Ooehara, J.; Hamanaka, S.; Onodera, M.; Rudolph, K.L.; Ema, H.; Nakauchi, H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell 2013, 154, 1112–1126. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Hu, W.; Naramura, M.; Park, C.Y. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J. Exp. Med. 2014, 211, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, D.G.; Copley, M.R.; Benz, C.; Wohrer, S.; Dykstra, B.J.; Ma, E.; Cheyne, J.; Zhao, T.; Gasparetto, M.; Delaney, M.; et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood 2009, 113, 6342–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansson, R.; Hultquist, A.; Luc, S.; Yang, L.; Anderson, K.; Kharazi, S.; Al-Hashmi, S.; Liuba, K.; Thoren, L.; Adolffsson, J.; et al. Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity 2007, 26, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Sanjuan-Pla, A.; Macaulay, I.C.; Jensen, C.T.; Woll, P.S.; Luis, T.C.; Mead, A.; Moore, S.; Carella, C.; Bouriez Jones, T.; Chowdhury, O.; et al. Platelet-biased stem cells reside at the apex of the hematopoietic stem-cell hierarchy. Nature 2013, 502, 232–236. [Google Scholar] [CrossRef]
- Luis, T.; Barkas, N.; Giustacchini, A.; Guerrero, J.; Wu, B.; Bouriez-Jones, T.; Macaulay, I.; Mead, A.; Nerlove, C.; Ghevaert, C.; et al. Perivascular niche cells sense thrombocytopenia and activate platelet-biased HSCs in an IL-1 dependent manner. Exp. Hematol. 2019, 76, S77. [Google Scholar] [CrossRef]
- Nutt, S.L.; Heavey, B.; Rolink, A.G.; Busslinger, M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 1999, 401, 556–562. [Google Scholar] [CrossRef]
- Rolink, A.G.; Nutt, S.L.; Melchers, F.; Busslinger, M. Long-term in vivo reconstitution of T-cell development by Pax5-deficient B-cell progenitors. Nature 1999, 401, 603–606. [Google Scholar] [CrossRef]
- Porritt, H.E.; Rumfelt, L.L.; Tabrizifard, S.; Schmitt, T.M.; Zuniga-Pflucker, J.C.; Petrie, H.T. Heterogeneity among DN1 prothymocytes reveals multiple progenitors with different capacities to generate T cell and non-T cell lineages. Immunity 2004, 20, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Balciunaite, G.; Ceredig, R.; Rolink, A.G. The earliest subpopulation of mouse thymocytes contains potent T, significant macrophage, and natural killer but no B-lymphocyte potential. Blood 2005, 105, 1930–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barik, S.; Cattin-Roy, A.N.; Miller, M.M.; Ukah, T.K.; Zaghouani, H. IL-4 and IL-13 guide early thymic progenitors to mature towards dendritic cells. J. Immunol. 2018, 201, 2945–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatman Zaretsky, A.; Taylor, J.J.; King, I.L.; Marshall, F.A.; Mohrs, M.; Pearce, E.J. T follicular helper cells differentiate from th2 cells in response to helminth antigens. J. Exp. Med. 2009, 206, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced cd18 levels drive regulatory t cell conversion into th17 cells in the cd18hypo pl/j mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.S.; Kim, I.K.; Park, Y.J.; Kim, Y.S.; Kim, Y.J.; Chang, W.S.; Lee, Y.S.; Kweon, M.N.; Chung, Y.; Kang, C.Y. Conversion of th2 memory cells into foxp3+ regulatory t cells suppressing th2-mediated allergic asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 8742–8747. [Google Scholar] [CrossRef] [Green Version]
- Eizenberg-Magar, I.; Rimer, J.; Zaretsky, I.; Lara-Astiaso, D.; Reich-Zeliger, S.; Friedman, N. Diverse continuum of cd4(+) t-cell states is determined by hierarchical additive integration of cytokine signals. Proc. Natl. Acad. Sci. USA 2017, 114, E6447–E6456. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory triggers associated with exacerbations of copd orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 2016, 17, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Shankland, M. Differentiation of the O and P cell lines in the embryo of the leech. Dev. Biol. 1987, 123, 97–107. [Google Scholar] [CrossRef]
- Moczek, A.P.; Sultan, S.; Foster, S.; Ledon-Rettig, C.; Dworkin, I.; Nijhout, H.F.; Abouheif, E.; Pfennig, D.W. The role of developmental plasticity in evolutionary innovation. Proc. R. Soc. B 2011, 278, 2705–2713. [Google Scholar] [CrossRef] [Green Version]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4808. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.; Hughes, P.J.; Ceredig, R. The versatile landscape of haematopoiesis: Are leukaemia stem cells as versatile? Crit. Rev. Clin. Lab. Sci. 2012, 49, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.F. Molecular genetics, natural history and the demise of childhood leukaemia. Eur. J. Cancer 1999, 35, 173–185. [Google Scholar] [CrossRef]
- Cox, C.V.; Blair, A. A primitive cell origin for B-cell precursor ALL? Stem Cell Rev. 2005, 1, 189–196. [Google Scholar] [CrossRef]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef]
- Couronne, L.; Bastard, C.; Bernard, O.A. TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 2012, 366, 95–96. [Google Scholar] [CrossRef]
- Weigert, O.; Weinstock, D.M. The evolving contribution of hematopoietic progenitor cells to lymphomagenesis. Blood 2012, 120, 2553–2561. [Google Scholar] [CrossRef]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Dueñas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.S.; Kim, E.; Park, J.H.; Chung, Y.R.; Lito, P.; Teruya-Feldstein, J.; Hu, W.; Beguelin, W.; Monette, S.; Duy, C.; et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci. Transl. Med. 2014, 6, 238ra271. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Enver, T. Acute promyelocytic leukaemia: Where does it stem from? Leukemia 2004, 18, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.H.; Wasik, M.A.; Finan, J.; Rodriguez, R.; Moore, J.; Kamoun, M.; Rennert, H.; Bird, J.; Nowell, P.C.; Salhany, K.E. Evidence for early hematopoietic progenitor cell involvement in acute promyelocytic leukemia. Am. J. Clin. Pathol. 1999, 112, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikushige, Y.; Miyamoto, T. Hematopoietic stem cell aging and chronic lymphocytic leukemia pathogenesis. Int. J. Hematol. 2014, 100, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sincennes, M.-C.; Humbert, M.; Grondin, B.; Lisi, V.; Veiga, D.F.T.; Haman, A.; Cazaux, C.; Mashtalir, N.; Affar, E.B.; Verreault, A.; et al. The LMO2 oncogene regulates DNA replication in hematopoietic cells. Proc. Natl. Acad. Sci. USA 2016, 113, 1393–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbitts, T.H. LMO T-cell translocation oncogenes typify genes activated by chromosomal translocations that alter transcription and developmental processes. Genes Dev. 1998, 12, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- McCormak, M.P.; Forster, A.; Drynaan, L.; Pannell, R.; Rabbitts, T.H. The LMO2 T-cell oncogene is activated via chromosomal translocations or retroviral insertion during gene therapy but has no mandatory role in normal T-cell development. Mol. Cell. Biol. 2003, 23, 9003–9013. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Warren, A.; Dobson, C.; Foster, A.; Pannell, R.; Rabbits, T.H. The T cell leukaemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc. Natl. Acad. Sci. USA 1998, 31, 3890–3895. [Google Scholar] [CrossRef] [Green Version]
- García-Ramírez, I.; Bhatia, S.; Rodríguez-Hernández, G.; González-Herrero, I.; Walter, C.; González de Tena-Dávila, S.; Parvin, S.; Haas, O.; Woessmann, W.; Stanulla, M.; et al. Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 2018, 37, e98783. [Google Scholar]
- Saglio, G.; Pane, F.; Martinelli, G.; Guerrasio, A. BCR/ABL rearrangement and leukemia phenotype. Leukemia 1999, 13, S96. [Google Scholar] [CrossRef] [Green Version]
- Ross, T.; Mgbemena, V.E. Re-evaluating the role of BCR/ABL in chronic myeloid leukaemia. Mol. Cell Oncol. 2014, 1, e963450. [Google Scholar] [CrossRef] [Green Version]
- Cazzaniga, G.; van Delft, F.W.; Nigro, L.O.; Ford, A.M.; Score, J.; Iacobucci, I.; Mirabile, E.; Tai, M.; Colman, S.M.; Biondi, A.; et al. Developmental origins and impact of BCR-ABL1 fusion and IKZF1 dleetions in monozygotic twins with Ph+ acute lymphoblastic leukaemia. Blood 2011, 118, 5559–5564. [Google Scholar] [CrossRef]
- Kosik, P.; Skorvaga, M.; Durdik, M.; Jakl, L.; Nikitina, E.; Markova, E.; Kozics, K.; Horvalthova, E.; Belyaev, I. Low numbers of pre-leukaemic fusion genes are frequently present in umbilical cord blood without affecting DNA damage response. Oncotarget 2017, 30, 35824–35834. [Google Scholar]
- Martín-Lorenzo, A.; Auer, F.; Chan, L.N.; García-Ramírez, I.; González-Herrero, I.; Rodríguez-Hernández, G.; Bartenhagen, C.; Dugas, M.; Gombert, M.; Ginzel, S.; et al. Loss of Pax5 Exploits Sca1-BCR-ABLp190 Susceptibility to Confer the Metabolic Shift Essential for pB-ALL. Cancer Res. 2018, 78, 2669–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Danes, A.; Blamplain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sánchez-Martín, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Dueñas, C.; González-Herrero, I.; Sehgal, L.; García-Ramírez, I.; Rodríguez-Hernández, G.; Pintado, B.; Blanco, O.; Criado, F.J.G.; Cenador, M.B.G.; Green, M.R.; et al. Dnm1 links BCR-ABLp210 to epigenetic tumour stem cell priming in myeloid leukaemia. Leukaemia 2019, 33, 249–278. [Google Scholar]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Stanulovic, V.S.; Cauchy, P.; Assi, S.A.; Hoogenkamp, M. LMO2 is required for TAL1 DNA binding activity and initiation of definitive haematopoiesis at the haemangioblast stage. Nucleic Acids Res. 2017, 45, 9874–9888. [Google Scholar] [CrossRef]
- Barndt, R.; Dai, M.; Zhuang, Y. Functions of E2A-HEB heterodimers in T-cell development revealed by a dominant negative mutant of HEB. Mol. Cell. Biol. 2000, 20, 6677–6685. [Google Scholar] [CrossRef] [Green Version]
- McCormack, M.P.; Young, L.F.; Vasudevan, S.; de Graaf, C.A.; Codrington, R.; Rabbitts, T.H.; Jane, S.M.; Curtis, D.J. The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 2010, 327, 879–883. [Google Scholar] [CrossRef]
- Wang, J.; Lee, S.; The, C.E.; Bunting, K.; Ma, L.; Shannon, M.F. The transcription repressor, ZEB11, cooperates with CtBP2 and HFAC1 to supress IL-2 gene activation in T-cells. Int. Immunol. 2009, 21, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Kaartinen, T.; Luostarinen, A.; Maliniemi, P.; Keto, J.; Arvas, M.; Belt, H.; Koponen, J.; Loskog, A.; Musdtjoki, S.; Porkka, K.; et al. Low interleukin-2 concentration favours generation of early memory T cells over effector phenotypes during chimeric antigen receptor T-cell expansion. Cytopathology 2017, 19, 689–702. [Google Scholar]
- Nakahata1, S.; Yamazaki, S.; Nakauchi, H.; Morishita, K. Downregulation of ZEB1 and overexpression of Smad7 contribute to resistance to TGF-β1-mediated growth suppression in adult T-cell leukemia/lymphoma. Oncogene 2010, 29, 4157–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Li, J.; Tian, C.; Shi, W.; Jiang, H.; Zhang, Z.; Wang, H.; Zhang, Q.; Sun, W.; Sun, P.; et al. Epigenetic dysregulation of ZEB1 is involved in LMO2-promoted T-cell acute lymphoblastic leukaemia leukaemogenesis. Biochem. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2511–2525. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, G.; Sánchez, L.; Sánchez-García, I. Lineage Decision-Making within Normal Haematopoietic and Leukemic Stem Cells. Int. J. Mol. Sci. 2020, 21, 2247. https://doi.org/10.3390/ijms21062247
Brown G, Sánchez L, Sánchez-García I. Lineage Decision-Making within Normal Haematopoietic and Leukemic Stem Cells. International Journal of Molecular Sciences. 2020; 21(6):2247. https://doi.org/10.3390/ijms21062247
Chicago/Turabian StyleBrown, Geoffrey, Lucía Sánchez, and Isidro Sánchez-García. 2020. "Lineage Decision-Making within Normal Haematopoietic and Leukemic Stem Cells" International Journal of Molecular Sciences 21, no. 6: 2247. https://doi.org/10.3390/ijms21062247
APA StyleBrown, G., Sánchez, L., & Sánchez-García, I. (2020). Lineage Decision-Making within Normal Haematopoietic and Leukemic Stem Cells. International Journal of Molecular Sciences, 21(6), 2247. https://doi.org/10.3390/ijms21062247