A Comparative Study of Engineered Dermal Templates for Skin Wound Repair in a Mouse Model

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
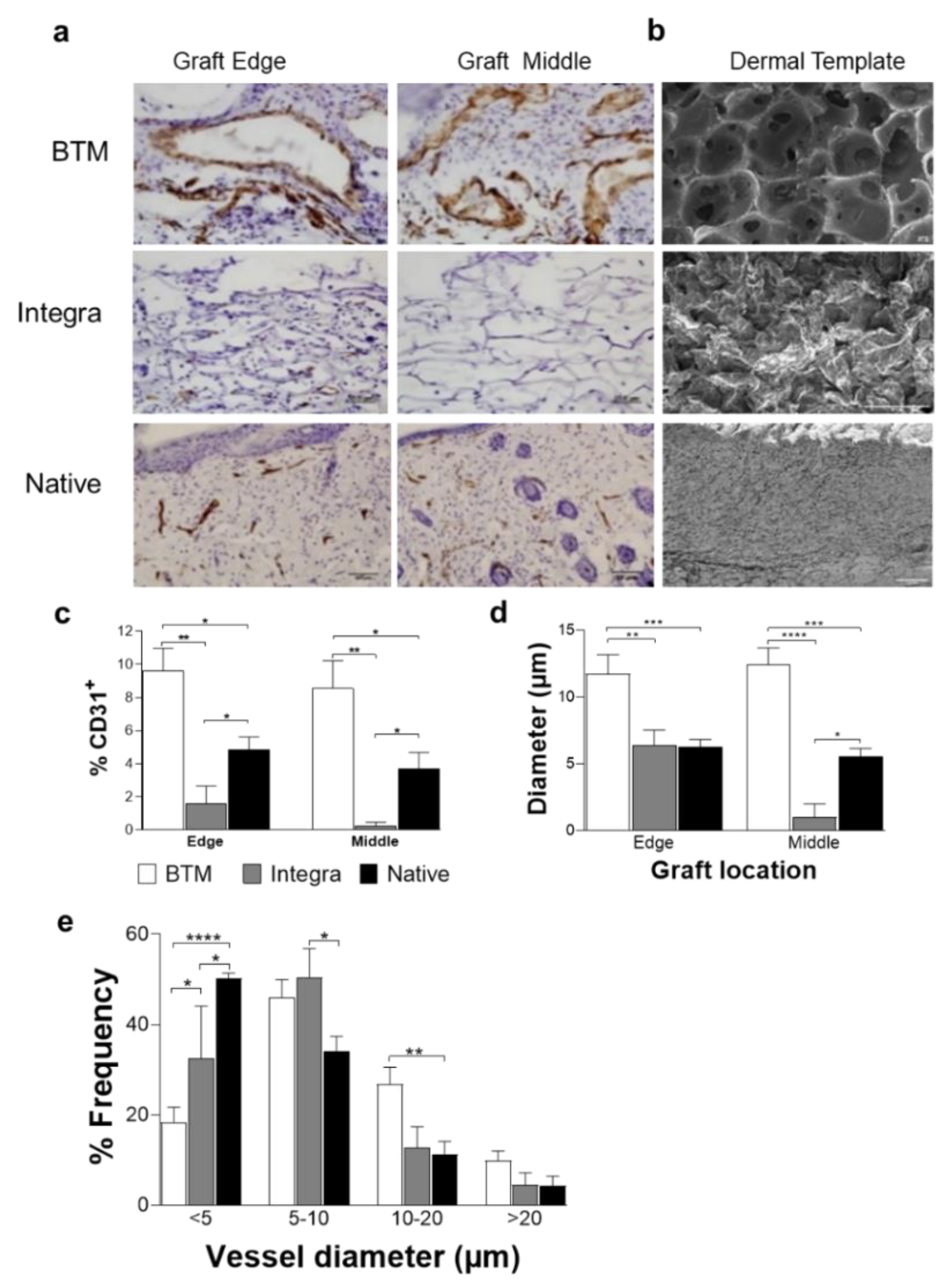
2.1. The Synthetic Dermal Template with Larger Pore Size Vascularises Faster than Collagen-Derived Dermal Template
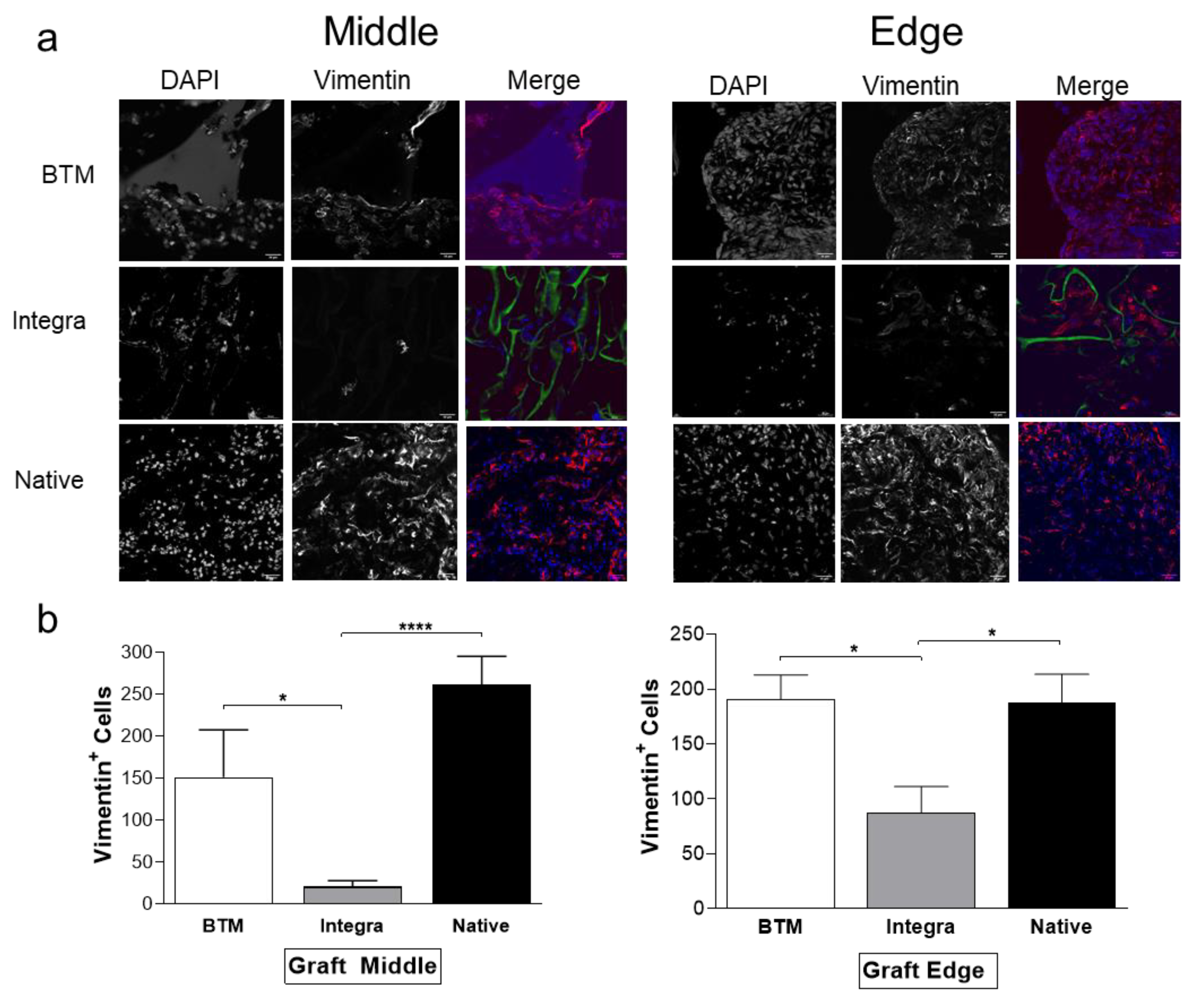
2.2. Host Dermal Fibroblast Infiltration Correlates with Vascularisation
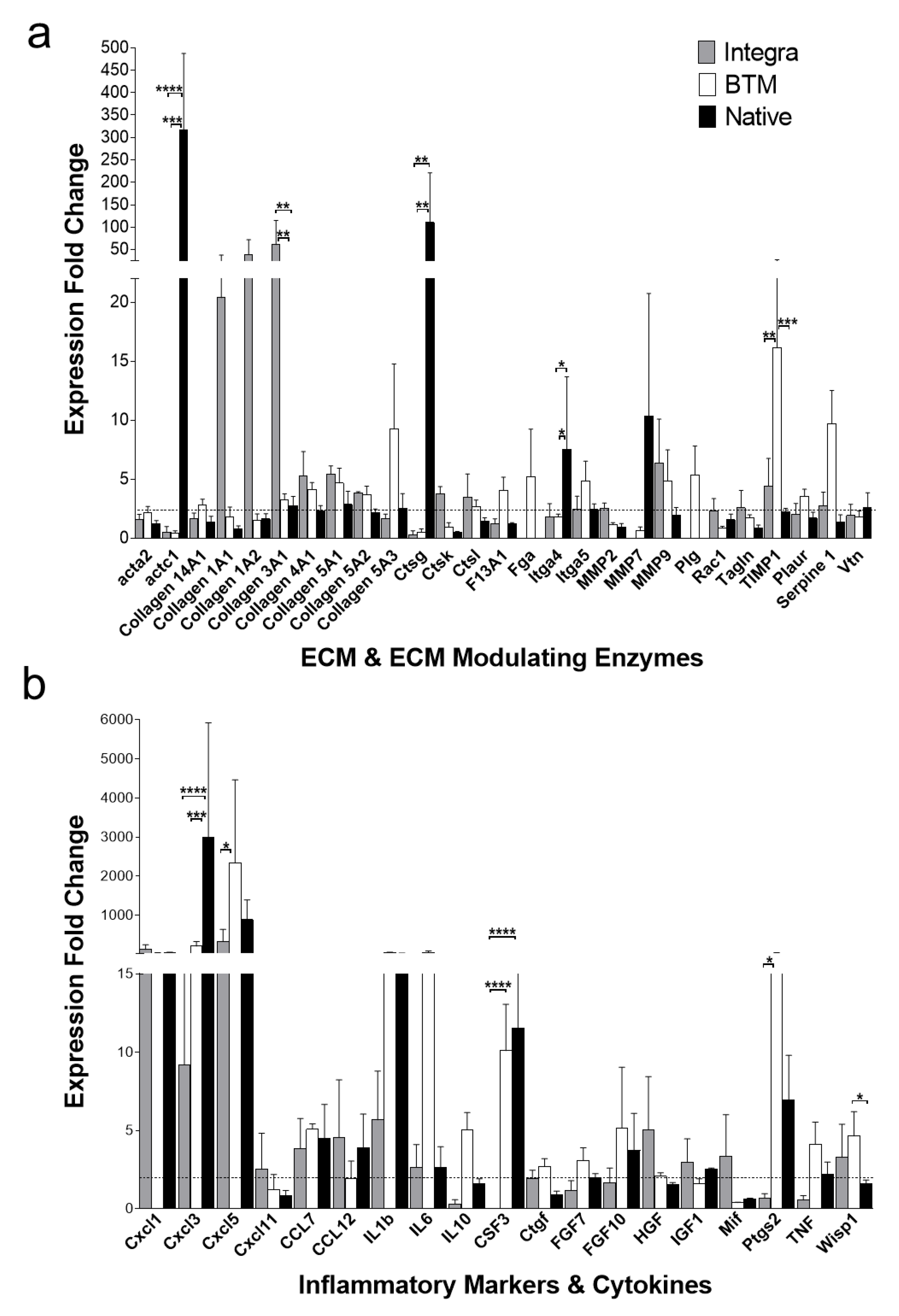
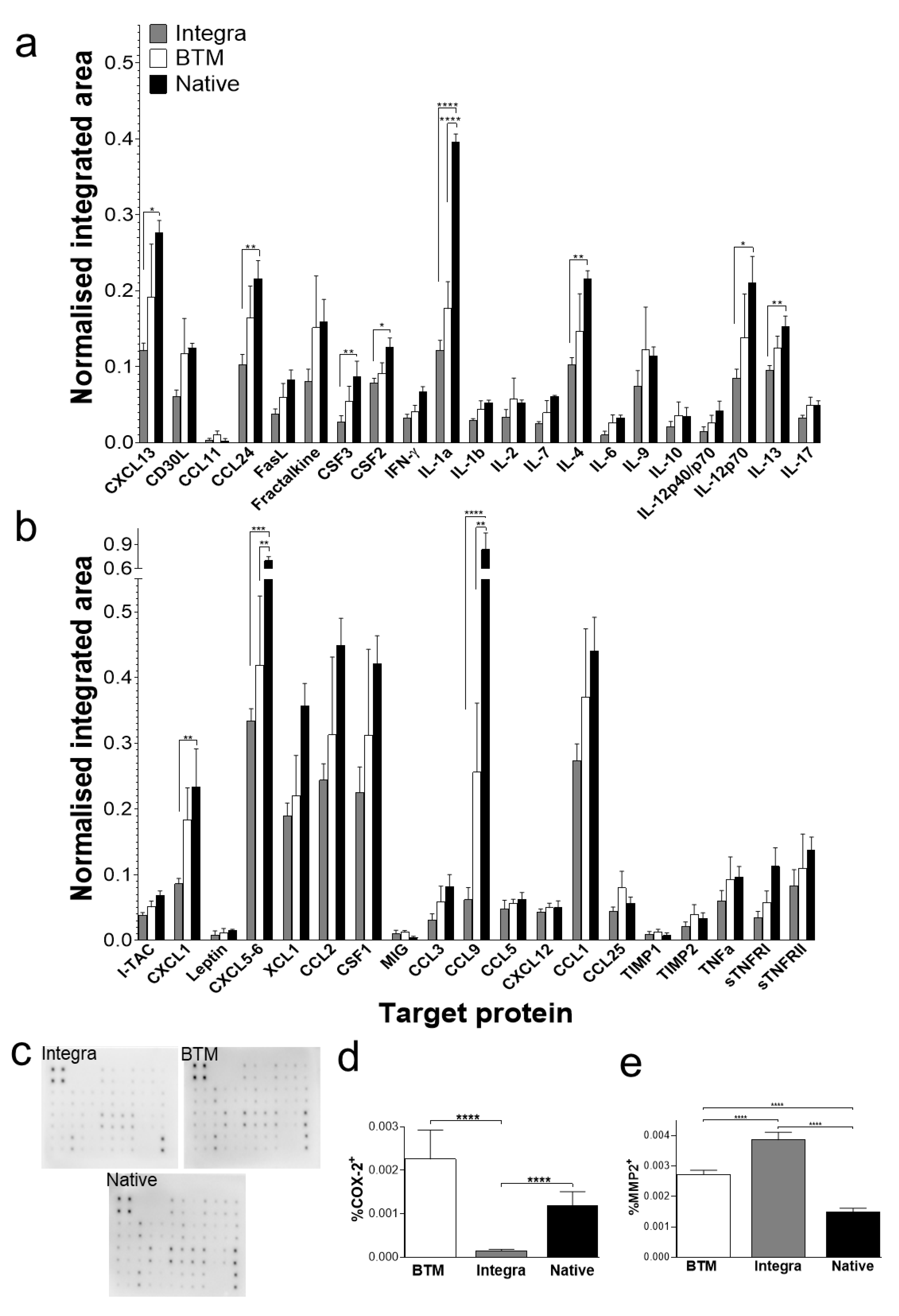
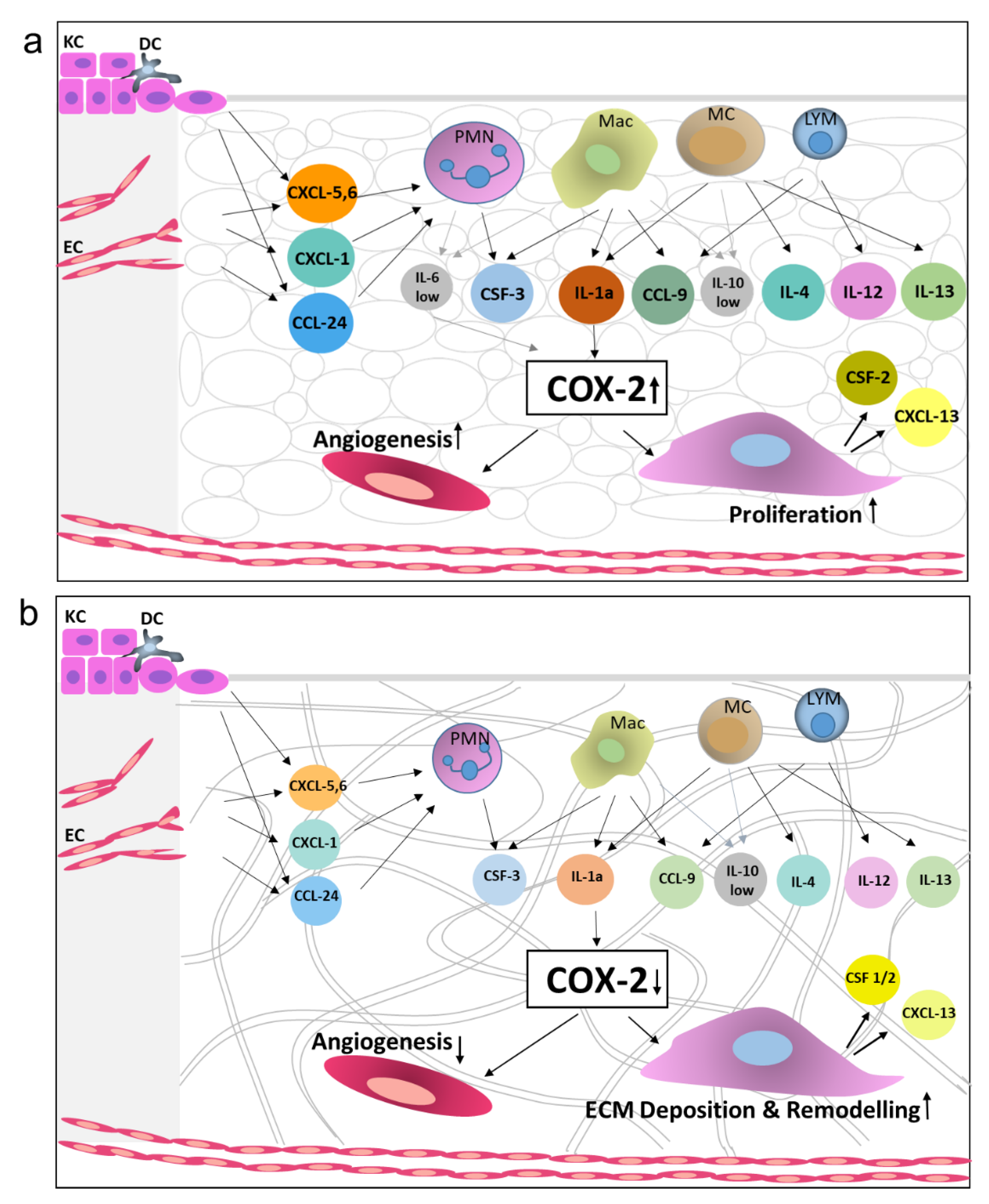
2.3. Identification of Molecular Mediators that May Influence Graft Take
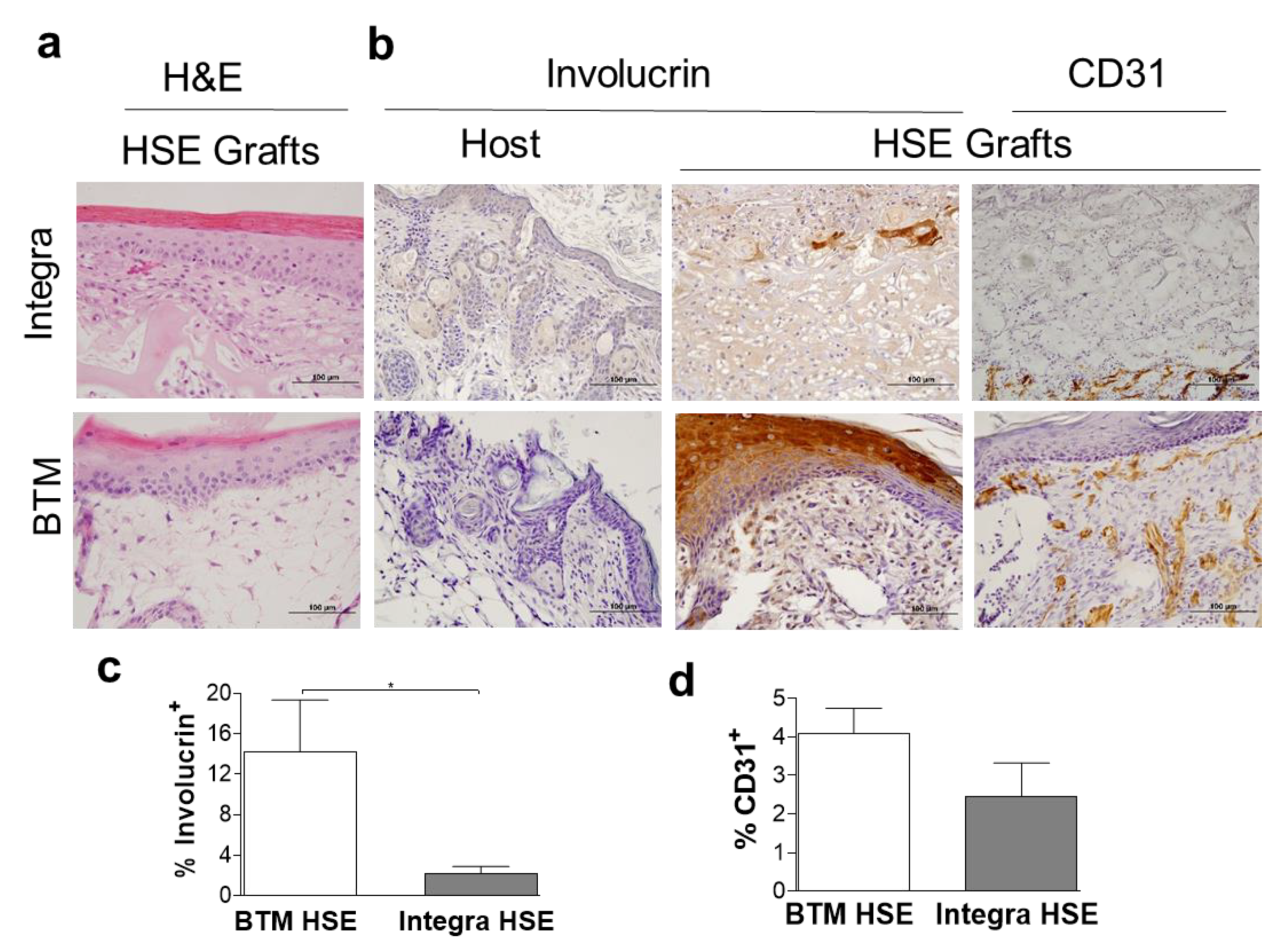
2.4. Application of Dermal Templates for Bioengineering a Human Skin Equivalent (HSE)
3. Discussion
4. Materials and Methods
4.1. Mouse Skin Grafting
4.2. Wound Healing Markers and ECM Expression Levels by Real Time PCR
4.3. Antibody Array
4.4. Immunohistochemistry and Analysis
4.5. Confocal Microscopy
4.6. Scanning Electron Microscopy
4.7. Human Skin Equivalent (HSE)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greenwood, J.E.; Dearman, B.L. Split skin graft application over an integrating, biodegradable temporizing polymer matrix: Immediate and delayed. J. Burn Care Res. Off. Publ. Am. Burn Assoc. 2012, 33, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.F.; Yannas, I.V.; Quinby, W.C., Jr.; Bondoc, C.C.; Jung, W.K. Successful use of a physiologically acceptable artificial skin in the treatment of extensive burn injury. Ann. Surg. 1981, 194, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Matsuda, K.; Maruguchi, T.; Nishimura, Y.; Ikada, Y. Further applications of “bilayer artificial skin”. Br. J. Plast. Surg. 1995, 48, 222–229. [Google Scholar] [CrossRef]
- Rygaard, J. Skin grafts in nude mice. 1. Allografts in nude mice of three genetic backgrounds (BALB-c, C3H, C57-BL). Acta Pathol. Microbiol. Scand. A 1974, 82, 80–92. [Google Scholar] [PubMed]
- Landen, N.X.; Li, D.; Stahle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell. Mol. Life Sci. 2016, 73, 3861–3885. [Google Scholar] [CrossRef] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Do, N.N.; Eming, S.A. Skin fibrosis: Models and mechanisms. Curr. Res. Transl. Med. 2016, 64, 185–193. [Google Scholar] [CrossRef]
- Wong, V.W.; Akaishi, S.; Longaker, M.T.; Gurtner, G.C. Pushing back: Wound mechanotransduction in repair and regeneration. J. Investig. Dermatol. 2011, 131, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Yannas, I.V.; Lee, E.; Orgill, D.P.; Skrabut, E.M.; Murphy, G.F. Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin. Proc. Natl. Acad. Sci. USA 1989, 86, 933–937. [Google Scholar] [CrossRef] [Green Version]
- Yannas, I.V. Facts and theories of induced organ regeneration. Adv. Biochem. Eng. Biotechnol. 2005, 93, 1–38. [Google Scholar]
- Lindenblatt, N.; Platz, U.; Althaus, M.; Hegland, N.; Schmidt, C.A.; Contaldo, C.; Vollmar, B.; Giovanoli, P.; Calcagni, M. Temporary angiogenic transformation of the skin graft vasculature after reperfusion. Plast. Reconstr. Surg. 2010, 126, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindstrom, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, J.M.; Breyer, M.D. Inflammatory modulation and wound repair. J. Investig. Dermatol. 2003, 120, xi–xii. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papacleovoulou, G.; Critchley, H.O.; Hillier, S.G.; Mason, J.I. IL1alpha and IL4 signalling in human ovarian surface epithelial cells. J. Endocrinol. 2011, 211, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Sphingosine-1-phosphate inhibits interleukin-1beta-induced inflammation in human articular chondrocytes. Int. J. Mol. Med. 2012, 30, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.; Kaur, P.; Herson, M.; Cheshire, P.; Cleland, H.; Akbarzadeh, S. Use of Clotted Human Plasma and Aprotinin in Skin Tissue Engineering: A Novel Approach to Engineering Composite Skin on a Porous Scaffold. Tissue Eng. Part C Methods 2015, 21, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Gillitzer, R.; Goebeler, M. Chemokines in cutaneous wound healing. J. Leukoc. Biol. 2001, 69, 513–521. [Google Scholar]
- Yukata, K.; Xie, C.; Li, T.F.; Brown, M.L.; Kanchiku, T.; Zhang, X.; Awad, H.A.; Schwarz, E.M.; Beck, C.A.; Jonason, J.H.; et al. Teriparatide (human PTH1-34) compensates for impaired fracture healing in COX-2 deficient mice. Bone 2018, 110, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, K.W.; Schellekens, D.H.; Bosmans, J.W.; Boonen, B.; Hulsewe, K.W.; Sastrowijoto, P.; Derikx, J.P.; Grootjans, J.; Poeze, M. Cyclooxygenase-2 Is Essential for Colorectal Anastomotic Healing. Ann. Surg. 2017, 265, 547–554. [Google Scholar] [CrossRef]
- Chikazu, D.; Fujikawa, Y.; Fujihara, H.; Suenaga, H.; Saijo, H.; Ohkubo, K.; Ogasawara, T.; Mori, Y.; Iino, M.; Takato, T. Cyclooxygenase-2 activity is important in craniofacial fracture repair. Int. J. Oral Maxillofac. Surg. 2011, 40, 322–326. [Google Scholar] [CrossRef]
- Geiser, T.; Dewald, B.; Ehrengruber, M.U.; Clark-Lewis, I.; Baggiolini, M. The interleukin-8-related chemotactic cytokines GRO alpha, GRO beta, and GRO gamma activate human neutrophil and basophil leukocytes. J. Biol. Chem. 1993, 268, 15419–15424. [Google Scholar] [PubMed]
- Tester, A.M.; Cox, J.H.; Connor, A.R.; Starr, A.E.; Dean, R.A.; Puente, X.S.; Lopez-Otin, C.; Overall, C.M. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS ONE 2007, 2, e312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaja-Milatovic, S.; Richmond, A. CXC chemokines and their receptors: A case for a significant biological role in cutaneous wound healing. Histol. Histopathol. 2008, 23, 1399–1407. [Google Scholar] [PubMed]
- Komi, D.E.A.; Khomtchouk, K.; Santa Maria, P.L. A Review of the Contribution of Mast Cells in Wound Healing: Involved Molecular and Cellular Mechanisms. Clin. Rev. Allergy Immunol. 2019, 58, 298–312. [Google Scholar] [CrossRef]
- Paquet, P.; Pierard, G.E. Interleukin-6 and the skin. Int. Arch. Allergy Immunol. 1996, 109, 308–317. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Elieh Ali Komi, D.; Ribatti, D. Mast cell-mediated mechanistic pathways in organ transplantation. Eur. J. Pharm. 2019, 857, 172458. [Google Scholar] [CrossRef]
- Gilroy, D.W.; Saunders, M.A.; Wu, K.K. COX-2 expression and cell cycle progression in human fibroblasts. Am. J. Physiol. Cell Physiol. 2001, 281, C188–C194. [Google Scholar] [CrossRef]
- Ahn, J.M.; Lee, J.S.; Um, S.G.; Rho, B.S.; Lee, K.B.; Park, S.G.; Kim, H.J.; Lee, Y.; Chi, Y.M.; Yoon, Y.E.; et al. Mussel adhesive Protein-conjugated Vitronectin (fp-151-VT) Induces Anti-inflammatory Activity on LPS-stimulated Macrophages and UVB-irradiated Keratinocytes. Immunol. Invest. 2019, 48, 242–254. [Google Scholar] [CrossRef]
- Hanna, N.; Bonifacio, L.; Weinberger, B.; Reddy, P.; Murphy, S.; Romero, R.; Sharma, S. Evidence for interleukin-10-mediated inhibition of cyclo- oxygenase-2 expression and prostaglandin production in preterm human placenta. Am. J. Reprod. Immunol. 2006, 55, 19–27. [Google Scholar] [CrossRef]
- Hol, J.; Wilhelmsen, L.; Haraldsen, G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J. Leukoc. Biol. 2010, 87, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, V.R.; Mintz, D.; Sagi, I. Matrix metalloproteinases: The sculptors of chronic cutaneous wounds. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt B, 2220–2227. [Google Scholar] [CrossRef]
- MacNeil, S. Biomaterials for tissue engineering of skin. Mater. Today 2008, 11, 26–35. [Google Scholar] [CrossRef]
- Gillard, J.A.; Reed, M.W.; Buttle, D.; Cross, S.S.; Brown, N.J. Matrix metalloproteinase activity and immunohistochemical profile of matrix metalloproteinase-2 and -9 and tissue inhibitor of metalloproteinase-1 during human dermal wound healing. Wound Repair Regen. 2004, 12, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Iba, Y.; Shibata, A.; Kato, M.; Masukawa, T. Possible involvement of mast cells in collagen remodeling in the late phase of cutaneous wound healing in mice. Int. Immunopharmacol. 2004, 4, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- Rippa, A.L.; Kalabusheva, E.P.; Vorotelyak, E.A. Regeneration of Dermis: Scarring and Cells Involved. Cells 2019, 8, 607. [Google Scholar] [CrossRef] [Green Version]
- MacNeil, S. Progress and opportunities for tissue-engineered skin. Nature 2007, 445, 874–880. [Google Scholar] [CrossRef]
- Bryan, D.; Walker, K.B.; Ferguson, M.; Thorpe, R. Cytokine gene expression in a murine wound healing model. Cytokine 2005, 31, 429–438. [Google Scholar] [CrossRef]
- Cheshire, P.A.; Herson, M.R.; Cleland, H.; Akbarzadeh, S. Artificial dermal templates: A comparative study of NovoSorb Biodegradable Temporising Matrix (BTM) and Integra((R)) Dermal Regeneration Template (DRT). Burns J. Int. Soc. Burn Inj. 2016, 42, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Corliss, B.A.; Mathews, C.; Doty, R.; Rohde, G.; Peirce, S.M. Methods to label, image, and analyze the complex structural architectures of microvascular networks. Microcirculation 2019, 26, e12520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheshire, P.; Zhafira, A.S.; Banakh, I.; Rahman, M.M.; Carmichael, I.; Herson, M.; Cleland, H.; Akbarzadeh, S. Xeno-free expansion of adult keratinocytes for clinical application: The use of human-derived feeder cells and serum. Cell Tissue Res. 2019, 376, 389–400. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banakh, I.; Cheshire, P.; Rahman, M.; Carmichael, I.; Jagadeesan, P.; Cameron, N.R.; Cleland, H.; Akbarzadeh, S. A Comparative Study of Engineered Dermal Templates for Skin Wound Repair in a Mouse Model. Int. J. Mol. Sci. 2020, 21, 4508. https://doi.org/10.3390/ijms21124508
Banakh I, Cheshire P, Rahman M, Carmichael I, Jagadeesan P, Cameron NR, Cleland H, Akbarzadeh S. A Comparative Study of Engineered Dermal Templates for Skin Wound Repair in a Mouse Model. International Journal of Molecular Sciences. 2020; 21(12):4508. https://doi.org/10.3390/ijms21124508
Chicago/Turabian StyleBanakh, Ilia, Perdita Cheshire, Mostafizur Rahman, Irena Carmichael, Premlatha Jagadeesan, Neil R. Cameron, Heather Cleland, and Shiva Akbarzadeh. 2020. "A Comparative Study of Engineered Dermal Templates for Skin Wound Repair in a Mouse Model" International Journal of Molecular Sciences 21, no. 12: 4508. https://doi.org/10.3390/ijms21124508
APA StyleBanakh, I., Cheshire, P., Rahman, M., Carmichael, I., Jagadeesan, P., Cameron, N. R., Cleland, H., & Akbarzadeh, S. (2020). A Comparative Study of Engineered Dermal Templates for Skin Wound Repair in a Mouse Model. International Journal of Molecular Sciences, 21(12), 4508. https://doi.org/10.3390/ijms21124508