The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling



Abstract
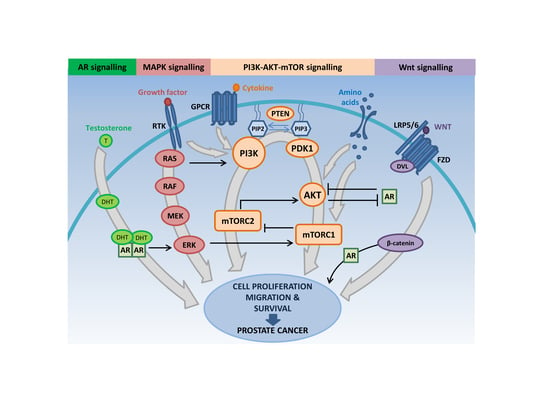
:
1. Introduction
2. Genetic Aberrations in the PI3K-AKT-mTOR Pathway in Prostate Cancer Are Diverse
2.1. PI3K Gain of Function
2.1.1. Class IA PI3Ks
2.1.2. Class IB PI3Ks
2.1.3. Class II PI3Ks
2.1.4. Class III PI3Ks
2.2. Loss of Function of Phosphoinositide Phosphatases
2.2.1. Loss or Inactivation of PTEN
2.2.2. Deregulation of Phosphoinositide Phosphatase Enzymes (other than PTEN)
2.3. AKT Gain of Function
2.3.1. AKT Mutation and Amplification
2.3.2. Genetic Alteration of AKT Regulators
2.4. SGK Deregulation
2.5. Loss of FOXO Transcription Factors
2.6. TSC1-TSC2-TBC1D7 Complex and RHEB Deregulation
2.7. Amplification of mTORC1 and mTORC2 Complex Components
2.8. Aberrant AMPK Signaling
2.8.1. CAMKKβ Amplification
2.8.2. LKB1 Loss
2.8.3. Sestrin Deletion
2.8.4. MAP3K7 Deletion
3. The PI3K-AKT-mTOR Pathway Intersects with Multiple Oncogenic Signaling Cascades to Facilitate Prostate Cancer Growth
3.1. PI3K-AKT-mTOR and RAS/MAPK Signaling Crosstalk
3.1.1. RAS/MAPK-PI3K-AKT-mTOR Interactions Promote Resistance to PI3K-AKT-mTOR Pathway-Directed Therapies
3.1.2. Co-targeting RAS/MAPK and PI3K-AKT-mTOR Signaling in Prostate Cancer
3.2. PI3K-AKT-mTOR and AR Signaling Crosstalk
3.3. PI3K-AKT-mTOR and WNT Signaling Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3β-HSD | 3β hydroxysteroid dehydrogenase |
| 4E-BP1 | Eukaryotic translation initiation factor 4E binding protein 1 |
| ADT | Androgen deprivation therapy |
| AD | Androstenedione |
| AGC | cAMP-dependent, cGMP-dependent and protein kinase C |
| AKTi | AKT inhibitor |
| AMP | Adenosine monophosphate |
| AMPK | 5′ AMP-activated protein kinase |
| APC | Adenomatous polyposis coli |
| AR | Androgen receptor |
| ARE | Androgen responsive element |
| ARF1 | Adenosine ribosylation factor 1 |
| ATG14 | Autophagy related 14 homolog |
| ATP | Adenosine triphosphate |
| AXIN | Axis inhibitor protein |
| BAD | Bcl-2-associated death promoter |
| BMK-1 | Big mitogen-activated protein kinase-1 |
| CaMKII | Calmodulin-dependent kinase 2 |
| CAMKKβ | Ca(2+)/calmodulin-dependent protein kinase kinase β |
| CAV1 | Caveolin-1 |
| CDK4 | Cyclin-dependent kinase 4 |
| c-JUN | Transcription factor AP-1 |
| CK1 | Casein kinase 1 |
| CAN | Copy number alteration |
| CREB | cAMP response element-binding protein |
| CRPC | Castrate resistant prostate cancer |
| CYP17A1 | Cytochrome P450 17A1 |
| DEPTOR | Dishevelled, EGL-10 and pleckstrin (DEP) domain-containing mTOR-interacting protein |
| DFCI | Dana-Farber Cancer Institute |
| DHEA | Dehydroepiandrosterone |
| DHTDVL | DihydrotestosteroneDisheveled |
| EIF4E | Eukaryotic translation initiation factor 4E |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EGR1 | Early growth response 1 |
| EMT | Epithelial-to-mesenchymal transition |
| ERG1 | ETS-related Gene 1 |
| ERK1 | Mitogen-activated protein kinase 3 |
| ERK2 | Mitogen-activated protein kinase 1 |
| EZH2 | Enhancer of zeste homolog 2 |
| FKBP5 | FK506 binding protein 5 |
| FOXO | Forkhead box protein O |
| FGFR | Fibroblast growth factor receptor |
| FZD | Frizzled family receptor |
| GAB1 | GRB2-associated binder-1 |
| GDP | Guanosine diphosphate |
| GLUT3 | Glucose transporter 3 |
| GnRH | Gonadotrophin-releasing hormone |
| GPCR | G-protein coupled receptor |
| GR | Glucocorticoid receptor |
| GRB2 | Growth factor receptor-bound protein 2 |
| GRB10 | Growth factor binding protein 10 |
| GSK3β | Glycogen synthase kinase 3 beta |
| GTP | Guanosine triphosphate |
| HDAC3 | Histone deacetylase 3 |
| HER2/3 | Human epidermal growth factor receptor 2/3 |
| HSD3B/17B3 | Hydroxysteroid dehydrogenase 3B/17B3 |
| HSP70/90 | Heat shock protein 70/90 |
| IGF | Insulin growth factor |
| IGF-1 | Insulin growth factor 1 |
| IHC | Immunohistochemistry |
| IL6 | Interleukin 6 |
| INPP4B | Inositol polyphosphate 4-phosphatase type II |
| INPP5D | Inositol polyphosphate-5-phosphatase D |
| INPP5J | Inositol polyphosphate-5-phosphatase J |
| INPPL1 | Inositol polyphosphate phosphatase like 1 |
| IRS | Insulin receptor substrate |
| IRS1 | Insulin receptor substrate protein 1 |
| KLK3 | Kallikrein related peptidase 3 |
| LAT1/2/3 | L-type amino acid transporter 1/2/3 |
| LDHA | L-lactate dehydrogenase A chain |
| LEF | Lymphoid enhancer binding factor |
| LH | Luteinizing hormone |
| LHRH | Luteinizing hormone-releasing hormone |
| LKB1 | Liver kinase B1 |
| LRP5/6 | Low-density lipoprotein receptor-related proteins 5 and 6 |
| MAPK | Mitogen-activated protein kinase |
| MAP3K7 | Mitogen-activated protein kinase kinase kinase 7 |
| mCRPC | Metastatic castrate resistant prostate cancer |
| MDM2 | Mouse double minute 2 homolog |
| MEK | Mitogen-activated protein kinase kinase |
| MSKCC | Memorial Sloan Kettering Cancer Centre |
| mLST8 | MTOR associated protein LST8 homolog |
| MMTV-PyMT | Mouse mammary tumor virus-polyoma middle tumor-antigen |
| MNK1 | MAP kinase interacting serine/threonine kinase 1 |
| MNK2 | MAP kinase interacting serine/threonine kinase 2 |
| mSIN1 | Mitogen-activated protein kinase associated protein 1 (MAPKAP1) |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mTORC1i | mTORC1 inhibitor |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| MuSK | Muscle specific kinase |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| NKX3.1 | NK3 Homeobox 1 |
| NSCLC | Non-small-cell lung carcinoma |
| P | Phosphorylation event |
| PDK1 | Phosphoinositide-dependent kinase 1 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PHLPP | PH domain leucine-rich repeat protein phosphatase |
| PHLPP1 | PH domain and leucine rich repeat protein phosphatase 1 |
| PHLPP2 | PH domain and leucine rich repeat protein phosphatase 2 |
| PI | Phosphatidylinositol |
| PI3K | Phosphoinositide 3-kinase |
| PI(3)P | Phosphatidylinositol 3-phosphate |
| PI(3,4)P2 | Phosphatidylinositol 3,4-bisphosphate |
| PIN | Prostate intra-epithelial neoplasia |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PIPP | Proline-rich inositol polyphosphate 5-phosphatase |
| PKB | Protein kinase B, AKT |
| PKC | Protein kinase |
| PKCα | Protein kinase C alpha |
| PLC | Phospholipase C |
| PP2A | Protein phosphatase 2A |
| PPP2CA | Protein phosphatase 2 catalytic subunit Alpha |
| PRAS40 | Proline-rich AKT substrate of 40 kDa |
| PROTOR1 | Protein observed with Rictor-1 |
| PROTOR2 | Protein observed with Rictor-2 |
| PSA | Prostate specific antigen |
| PTEN | Phosphatase and tensin homologue deleted on chromosome 10 |
| PTK7 | Protein tyrosine kinase 7 |
| RAF | Rapidly accelerated fibrosarcoma |
| RAG | Recombination activating genes |
| RAPTOR | Regulatory-associated protein of mTOR |
| RBD | Ras-binding domain |
| RHEB | Ras homolog enriched in brain |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| ROR1/2 | Receptor tyrosine kinase–like orphan receptor-1 and -2 |
| RPS6 | Ribosomal protein S6 |
| rPFS | Radiographic progression-free survival |
| RSK | 90 kDa Ribosomal S6 kinase |
| RTK | Tyrosine kinase receptor |
| RYK | Receptor-like tyrosine kinase |
| S6K | Ribosomal protein S6 kinase/p70 ribosomal S6 kinase |
| SESN1 | Sestrin 1 |
| SGK1 | Serum and glucocorticoid regulated kinase 1 |
| SGK2 | Serum and glucocorticoid regulated kinase 2 |
| SGK3 | Serum and glucocorticoid regulated kinase 3 |
| SH2 | Src homology 2 |
| SHIP1 | SH2 domain-containing inositol 5′-phosphatase 1 |
| SHIP2 | SH2 domain-containing inositol 5′-Phosphatase 2 |
| SLC2A3 | Solute carrier family 2, facilitated glucose transporter member 3 |
| SLC43A1 | Solute carrier family 43 member 1 |
| SOS | Son of Sevenless |
| SPOP | Speckle type BTB/POZ protein |
| SRC-3 | Steroid receptor co-activator 3 |
| SRF | Serum response factor |
| SU2C-PCF IDT | Stand Up To Cancer & Prostate Cancer Foundation International Dream Team |
| T | Testosterone |
| TAK1 | TGFβ-activated kinase 1 |
| TAZ | Transcriptional coactivator with PDZ binding motif |
| TBC1D7 | TBC1 Domain Family Member 7 |
| TCF | T cell Factor |
| TCGA | The Cancer Genome Atlas |
| TEAD | Transcriptional enhanced associate domain |
| TEL2 | Telomere length regulation protein |
| TGF | Transforming growth factor |
| TMPRSS2-ERG | Transmembrane protease, serine 2: ETS Transcription Factor fusion |
| TRAMP | Transgenic adenocarcinoma mouse prostate |
| TSC1 | Tuberous Sclerosis complex 1 |
| TSC2 | Tuberous Sclerosis complex 2 |
| TTI1 | TELO2 interacting protein 1 |
| Ub | Ubiquitination event |
| ULK1 | Unc-51 Like Autophagy Activating Kinase 1 |
| UVRAG | UV radiation resistance-associated gene; v-ATPase, Vacuolar (H+)-ATPase |
| VPS15 | Vacuolar protein sorting 15 |
| V-ATPase | Vacuolar H+-ATPase |
| WNT | WNT ligand |
| YAP | Yes-associated protein |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, N.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, E359–E386. [Google Scholar] [CrossRef]
- Jemal, A.; Fedewa, S.A.; Ma, J.; Siegel, R.; Lin, C.C.; Brawley, O.; Ward, E.M. Prostate Cancer Incidence and PSA Testing Patterns in Relation to USPSTF Screening Recommendations. JAMA 2015, 314, 2054–2061. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.B.; Li, J.; Huang, B.; Weir, H.K. Prostate cancer survival in the United States by race and stage (2001–2009): Findings from the CONCORD-2 study. Cancer 2017, 123, 5160–5177. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer treatment and survivorship statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prostate Cancer Prostatic Dis. 2018, 22, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culig, Z. Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr. Mol. Biol. Rep. 2017, 3, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacinti, S.; Bassanelli, M.; Aschelter, A.M.; Milano, A.; Roberto, M.; Marchetti, P. Resistance to abiraterone in castration-resistant prostate cancer: A review of the literature. Anticancer Res. 2014, 34, 6265–6269. [Google Scholar] [PubMed]
- Rice, M.A.; Malhotra, S.V.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801. [Google Scholar] [CrossRef]
- Perlmutter, M.A.; Lepor, H. Androgen Deprivation Therapy in the Treatment of Advanced Prostate Cancer. Rev. Urol. 2007, 9, S3–S8. [Google Scholar]
- Mostaghel, E.A. Abiraterone in the treatment of metastatic castration-resistant prostate cancer. Cancer Manag. Res. 2014, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.; Li, J.; Méniel, V.; Fennell, C.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; MacPherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat. Cancer 2013, 20, R83–R99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, A.C.; Edlind, M.P. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities (Review). Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K Pathway as Drug Target in Human Cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Thorpe, L.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2014, 15, 7–24. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillermet-Guibert, J.; Bjorklof, K.; Salpekar, A.; Gonella, C.; Ramadani, F.; Bilancio, A.; Meek, S.; Smith, A.J.H.; Okkenhaug, K.; Vanhaesebroeck, B. The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc. Natl. Acad. Sci. USA 2008, 105, 8292–8297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Pandolfi, P.P. The PTEN-PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2018, 48, 27–35. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Berenjeno, I.M.; Piñeiro, R.; Castillo, S.D.; Pearce, W.; McGranahan, N.; Dewhurst, S.M.; Meniel, V.; Birkbak, N.J.; Lau, E.; Sansregret, L.; et al. Oncogenic PIK3CA induces centrosome amplification and tolerance to genome doubling. Nat. Commun. 2017, 8, 1773. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Julien, L.-A.; Carrière, A.; Moreau, J.; Roux, P.P. mTORC1-Activated S6K1 Phosphorylates Rictor on Threonine 1135 and Regulates mTORC2 Signaling. Mol. Cell. Biol. 2009, 30, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C.; Wajant, H. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, K.; Mahajan, N.P. PI3K-independent AKT activation in cancers: A treasure trove for novel therapeutics. J. Cell. Physiol. 2012, 227, 3178–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faes, S.; Faes, O.D.S. PI3K and AKT: Unfaithful Partners in Cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Dibble, C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Jewell, J.L.; Russell, R.C.; Guan, K.-L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.N.; Brattain, M.; Ghosh, P.M.; Troyer, D.A.; Prihoda, T.; Bedolla, R.; Kreisberg, J.I. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin. Cancer Res. 2002, 8, 1168–1171. [Google Scholar]
- Kremer, C.L.; Klein, R.R.; Mendelson, J.; Browne, W.; Samadzedeh, L.K.; Vanpatten, K.; Highstrom, L.; Pestano, G.A.; Nagle, R. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate 2006, 66, 1203–1212. [Google Scholar] [CrossRef]
- Sutherland, S.I.; Benito, R.P.; Henshall, S.M.; Horvath, L.; Kench, J.G. Expression of phosphorylated-mTOR during the development of prostate cancer. Prostate 2014, 74, 1231–1239. [Google Scholar] [CrossRef]
- Liao, Y.; Grobholz, R.; Abel, U.; Trojan, L.; Michel, M.S.; Angel, P.; Mayer, D. Increase of AKT/PKB expression correlates with gleason pattern in human prostate cancer. Int. J. Cancer 2003, 107, 676–680. [Google Scholar] [CrossRef]
- Evren, S.; Dermen, A.; Lockwood, G.; Fleshner, N.; Sweet, J. mTOR–RAPTOR and 14-3-3? immunohistochemical expression in high grade prostatic intraepithelial neoplasia and prostatic adenocarcinomas: A tissue microarray study. J. Clin. Pathol. 2011, 64, 683–688. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Skanderup, A.J.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Poulogiannis, G.; Pyne, S.; Jia, S.; Zou, L.; Signoretti, S.; Loda, M.; Cantley, L.C.; Roberts, T.M. A constitutively activated form of the p110β isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 11002–11007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Outmezguine, V.R.-; Will, M.; Yellen, P.; De Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell 2014, 27, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Gao, X.; Lee, S.H.; Maira, S.-M.; Wu, X.; Stack, E.C.; Signoretti, S.; Loda, M.; Zhao, J.J.; Roberts, T.M. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2012, 3, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.C.; Warne, P.H.; Zvelebil, M.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. p110, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef] [Green Version]
- Chantry, D.; Vojtek, A.; Kashishian, A.; Holtzman, D.A.; Wood, C.; Gray, P.W.; Cooper, J.A.; Hoekstra, M.F. p110δ, a Novel Phosphatidylinositol 3-Kinase Catalytic Subunit That Associates with p85 and Is Expressed Predominantly in Leukocytes. J. Biol. Chem. 1997, 272, 19236–19241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eickholt, B.J.; Ahmed, A.; Davies, M.; Papakonstanti, E.; Pearce, W.; Starkey, M.L.; Bilancio, A.; Need, A.C.; Smith, A.J.H.; Hall, S.M.; et al. Control of Axonal Growth and Regeneration of Sensory Neurons by the p110δ PI 3-Kinase. PLoS ONE 2007, 2, e869. [Google Scholar] [CrossRef] [Green Version]
- Tzenaki, N.; Papakonstanti, E. p110δ PI3 kinase pathway: Emerging roles in cancer. Front. Oncol. 2013, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Tzenaki, N.; Andreou, M.; Stratigi, K.; Vergetaki, A.; Makrigiannakis, A.; Vanhaesebroeck, B.; Papakonstanti, E. High levels of p110δ PI3K expression in solid tumor cells suppress PTEN activity, generating cellular sensitivity to p110δ inhibitors through PTEN activation. FASEB J. 2012, 26, 2498–2508. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.; Syed, A.; Middha, S.; Kim, H.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Wang, B.-D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef]
- Ueki, K.; Yballe, C.M.; Brachmann, S.M.; Vicent, D.; Watt, J.M.; Kahn, C.R.; Cantley, L.C. Increased insulin sensitivity in mice lacking p85 subunit of phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2001, 99, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Field, S.J.; Lee, J.Y.; Engelman, J.A.; Cantley, L.C. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J. Cell Biol. 2005, 170, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Terauchi, Y.; Tsuji, Y.; Satoh, S.; Minoura, H.; Murakami, K.; Okuno, A.; Inukai, K.; Asano, T.; Kaburagi, Y.; Ueki, K.; et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85α subunit of phosphoinositide 3–kinase. Nat. Genet. 1999, 21, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Mauvais-Jarvis, F.; Ueki, K.; Fruman, D.A.; Hirshman, M.F.; Sakamoto, K.; Goodyear, L.J.; Iannacone, M.; Accili, M.; Cantley, L.C.; Kahn, C.R. Reduced expression of the murine p85α subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J. Clin. Investig. 2002, 109, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Winnay, J.; Kondo, T.; Bronson, R.T.; Guimaraes, A.R.; Aleman, J.O.; Luo, J.; Stephanopoulos, G.; Weissleder, R.; Cantley, L.C.; et al. The phosphoinositide 3-kinase regulatory subunit p85alpha can exert tumor suppressor properties through negative regulation of growth factor signaling. Cancer Res. 2010, 70, 5305–5315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorpe, L.M.; Spangle, J.M.; Ohlson, C.E.; Cheng, H.; Roberts, T.M.; Cantley, L.C.; Zhao, J.J. PI3K-p110α mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85α. Proc. Natl. Acad. Sci. USA 2017, 114, 7095–7100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3’-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar] [PubMed]
- Vallejo-Díaz, J.; Chagoyen, M.; Olazabal-Morán, M.; Gonzalez-Garcia, A.; Carrera, A. The Opposing Roles of PIK3R1/p85α and PIK3R2/p85β in Cancer. Trends Cancer 2019, 5, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Diaz, J.; Olazabal-Morán, M.; Cariaga-Martinez, A.E.; Pajares, M.J.; Flores, J.M.; Pio, R.; Montuenga, L.M.; Carrera, A. Targeted depletion of PIK3R2 induces regression of lung squamous cell carcinoma. Oncotarget 2016, 7, 85063–85078. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Huang, J.; Yang, N.; Greshock, J.; Liang, S.; Hasegawa, K.; Giannakakis, A.; Poulos, N.; O’Brien-Jenkins, A.; Katsaros, D.; et al. Integrative genomic analysis of phosphatidylinositol 3’-kinase family identifies PIK3R3 as a potential therapeutic target in epithelial ovarian cancer. Clin. Cancer Res. 2007, 13, 5314–5321. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.-P.; Zhu, Y.; Yin, L.-D.; Wei, J.-S.; Liu, X.-C.; Zhu, X.-L.; Miao, Y. PIK3R3 Promotes Metastasis of Pancreatic Cancer via ZEB1 Induced Epithelial-Mesenchymal Transition. Cell. Physiol. Biochem. 2018, 46, 1930–1938. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J.; Livermore, K.; McClurgg, U.L.; Kalna, G.; Knight, B.; Mccullagh, P.; McGrath, J.; Crundwell, M.; Leung, H.Y.; Robson, C.; et al. The PI3K regulatory subunit gene PIK3R1 is under direct control of androgens and repressed in prostate cancer cells. Oncoscience 2015, 2, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Zhang, L.; Gan, W.; Fu, K.; Jiang, K.; Ding, J.; Wu, J.; Han, X.; Li, D. piRNA-DQ722010 contributes to prostate hyperplasia of the male offspring mice after the maternal exposed to microcystin-leucine arginine. Prostate 2019, 79, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Xie, X.; Yu, S.; Peng, F.; Peng, L. MicroRNA-126 inhibits proliferation and metastasis by targeting pik3r2 in prostate cancer. Mol. Med. Rep. 2015, 13, 1204–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazzatti, J.; Klingler-Hoffmann, M.; Haylock-Jacobs, S.; Harata-Lee, Y.; Niu, M.; Higgins, M.D.; Kochetkova, M.; Hoffmann, P.; McColl, S.R. Differential roles for the p101 and p84 regulatory subunits of PI3Kγ in tumor growth and metastasis. Oncogene 2011, 31, 2350–2361. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, K.; Hammerman, P.S.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.F.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e6. [Google Scholar] [CrossRef] [Green Version]
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Mavrommati, I.; Cisse, O.; Falasca, M.; Maffucci, T. Novel roles for class II Phosphoinositide 3-Kinase C2β in signalling pathways involved in prostate cancer cell invasion. Sci. Rep. 2016, 6, 23277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiborg, C.; Schink, K.O.; Stenmark, H.A. Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J. 2013, 280, 2730–2742. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Alliouachene, S.; Pearce, W.; Morelli, D.; Szabadkai, G.; Chung, Y.-L.; Chicanne, G.; Valet, C.; Hill, J.M.; Voshol, P.J.; et al. Vps34 PI 3-kinase inactivation enhances insulin sensitivity through reprogramming of mitochondrial metabolism. Nat. Commun. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef] [Green Version]
- Nezis, I.P.; Sagona, A.P.; Schink, K.O.; Stenmark, H. Divide and ProsPer: The emerging role of PtdIns3P in cytokinesis. Trends Cell Biol. 2010, 20, 642–649. [Google Scholar] [CrossRef]
- Yoon, M.-S.; Son, K.; Arauz, E.; Han, J.M.; Kim, S.; Chen, J. Leucyl-tRNA Synthetase Activates Vps34 in Amino Acid-Sensing mTORC1 Signaling. Cell Rep. 2016, 16, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dyson, J.M.; Fedele, C.G.; Davies, E.M.; Becanovic, J.; Mitchell, C.A. Phosphoinositide Phosphatases: Just as Important as the Kinases. Subcell. Biochem. 2012, 58, 215–279. [Google Scholar] [CrossRef]
- Rudge, S.A.; Wakelam, M.J. Phosphatidylinositolphosphate phosphatase activities and cancer. J. Lipid Res. 2015, 57, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Cairns, P.; Okami, K.; Halachmi, S.; Halachmi, N.; Esteller, M.; Herman, J.G.; Jen, J.; Isaacs, W.B.; Bova, G.S.; Sidransky, D. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997, 57, 4997–5000. [Google Scholar] [PubMed]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar] [PubMed]
- Wang, S.I.; Parsons, R.; Ittmann, M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin. Cancer Res. 1998, 4, 811–815. [Google Scholar] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, M.P.; Stolarov, J.P.; Eng, C.; Li, J.; Wang, S.I.; Wigler, M.; Parsons, R.; Tonks, N.K. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. USA 1997, 94, 9052–9057. [Google Scholar] [CrossRef] [Green Version]
- Geybels, M.S.; Fang, M.; Wright, J.L.; Qu, X.; Bibikova, M.; Klotzle, B.; Fan, J.-B.; Feng, Z.; Ostrander, E.A.; Nelson, P.S.; et al. PTEN loss is associated with prostate cancer recurrence and alterations in tumor DNA methylation profiles. Oncotarget 2017, 8, 84338–84348. [Google Scholar] [CrossRef] [Green Version]
- McMenamin, M.E.; Soung, P.; Perera, S.; Kaplan, I.; Loda, M.; Sellers, W.R. Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res. 1999, 59, 4291–4296. [Google Scholar]
- Ratnacaram, C.K.; Telentin, M.; Jiang, M.; Meng, X.; Chambon, P.; Metzger, D. Temporally controlled ablation of PTEN in adult mouse prostate epithelium generates a model of invasive prostatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 2521–2526. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Gao, J.; Lei, Q.-Y.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, I.; Patel, R.; Singh, L.B.; Nixon, C.; Seywright, M.; Barnetson, R.J.; Brunton, V.G.; Muller, W.J.; Edwards, J.; Sansom, O.J.; et al. HER2 overcomes PTEN (loss)-induced senescence to cause aggressive prostate cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 16392–16397. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.K.; Johnson, D.T.; Zhu, C.; Lee, S.H.; Ye, D.-W.; Luong, R.; Sun, Z. Conditional Deletion of the Pten Gene in the Mouse Prostate Induces Prostatic Intraepithelial Neoplasms at Early Ages but a Slow Progression to Prostate Tumors. PLoS ONE 2013, 8, e53476. [Google Scholar] [CrossRef]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003, 1, e59. [Google Scholar] [CrossRef]
- Manda, K.R.; Tripathi, P.; Hsi, A.C.; Ning, J.; Ruzinova, M.B.; Liapis, H.; Bailey, M.; Zhang, H.; Maher, C.A.; Humphrey, P.A.; et al. NFATc1 promotes prostate tumorigenesis and overcomes PTEN loss-induced senescence. Oncogene 2015, 35, 3282–3292. [Google Scholar] [CrossRef] [Green Version]
- Han, S.Y.; Kato, H.; Kato, S.; Suzuki, T.; Shibata, H.; Ishii, S.; Shiiba, K.; Matsuno, S.; Kanamaru, R.; Ishioka, C. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 2000, 60, 3147–3151. [Google Scholar]
- Correia, N.C.; Gírio, A.; Antunes, I.; Martins, L.R.; Barata, J. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur. J. Cancer 2014, 50, 216–225. [Google Scholar] [CrossRef]
- Ooms, L.M.; Binge, L.C.; Davies, E.M.; Rahman, P.; Conway, J.R.; Gurung, R.; Ferguson, D.T.; Papa, A.; Fedele, C.G.; Vieusseux, J.L.; et al. The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell 2015, 28, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, S.J.; Ferguson, D.T.; Mitchell, C.A.; Ooms, L.M. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewinner, C.; Wang, Z.C.; Richardson, A.; Teruya-Feldstein, J.; Etemadmoghadam, D.; Bowtell, D.D.; Barretina, J.; Lin, W.M.; Rameh, L.; Salmena, L.; et al. Evidence that Inositol Polyphosphate 4-Phosphatase Type II Is a Tumor Suppressor that Inhibits PI3K Signaling. Cancer Cell 2009, 16, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgson, M.C.; Shao, L.-J.; Frolov, A.; Li, R.; Peterson, L.; Ayala, G.; Ittmann, M.M.; Weigel, N.L.; Agoulnik, I.U. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011, 71, 572–582. [Google Scholar] [CrossRef] [Green Version]
- Fedele, C.G.; Ooms, L.M.; Ho, M.; Vieusseux, J.; O’Toole, S.A.; Millar, E.; Knowles, E.L.; Sriratana, A.; Gurung, R.; Baglietto, L.; et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl. Acad. Sci. USA 2010, 107, 22231–22236. [Google Scholar] [CrossRef] [Green Version]
- Kofuji, S.; Kimura, H.; Nakanishi, H.; Nanjo, H.; Takasuga, S.; Liu, H.; Eguchi, S.; Nakamura, R.; Itoh, R.; Ueno, N.; et al. INPP4B Is a PtdIns(3,4,5)P3 Phosphatase That Can Act as a Tumor Suppressor. Cancer Discov. 2015, 5, 730–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, C.L.; Lunardi, A.; Gulluni, F.; Ruan, D.T.; Chen, M.; Salmena, L.; Nishino, M.; Papa, A.; Ng, C.; Fung, J.; et al. In Vivo Role of INPP4B in Tumor and Metastasis Suppression through Regulation of PI3K-AKT Signaling at Endosomes. Cancer Discov. 2015, 5, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, H.; Chen, Q. INPP4B overexpression suppresses migration, invasion and angiogenesis of human prostate cancer cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 700–708. [Google Scholar] [CrossRef]
- Rynkiewicz, N.K.; Fedele, C.G.; Chiam, K.; Gupta, R.; Kench, J.G.; Ooms, L.M.; McLean, C.; Giles, G.G.; Horvath, L.; Mitchell, C.A. INPP4B is highly expressed in prostate intermediate cells and its loss of expression in prostate carcinoma predicts for recurrence and poor long term survival. Prostate 2014, 75, 92–102. [Google Scholar] [CrossRef]
- Xie, J.; Erneux, C.; Pirson, I. How does SHIP1/2 balance PtdIns(3,4)P 2 and does it signal independently of its phosphatase activity? BioEssays 2013, 35, 733–743. [Google Scholar] [CrossRef]
- Hoekstra, E.; Das, A.M.; Willemsen, M.; Swets, M.; Kuppen, P.J.; Van Der Woude, C.J.; Bruno, M.J.; Shah, J.P.; Hagen, T.L.T.; Chisholm, J.D.; et al. Lipid phosphatase SHIP2 functions as oncogene in colorectal cancer by regulating PKB activation. Oncotarget 2016, 7, 73525–73540. [Google Scholar] [CrossRef]
- Chan, T.O.; Rittenhouse, S.E.; Tsichlis, P.N. AKT/PKB and Other D3 Phosphoinositide-Regulated Kinases: Kinase Activation by Phosphoinositide-Dependent Phosphorylation. Annu. Rev. Biochem. 1999, 68, 965–1014. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; Deak, M.; Casamayor, A.; Caudwell, F.B.; Morrice, N.; Norman, D.G.; Gaffney, P.; Reese, C.B.; MacDougall, C.N.; Harbison, D.; et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): Structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol. 1997, 7, 776–789. [Google Scholar] [CrossRef] [Green Version]
- Burgering, B.; Coffer, P.J. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995, 376, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Andjelković, M.; Jakubowicz, T.; Cron, P.; Ming, X.F.; Han, J.W.; Hemmings, B.A. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proc. Natl. Acad. Sci. USA 1996, 93, 5699–5704. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Furnari, F.; Newton, A.C. PHLPP: A Phosphatase that Directly Dephosphorylates Akt, Promotes Apoptosis, and Suppresses Tumor Growth. Mol. Cell 2005, 18, 13–24. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.-J.; et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Felicioni, L.; Buttitta, F.; Lamba, S.; Cardone, L.; Rodolfo, M.; Scarpa, A.; Leenstra, S.; Frattini, M.; Barbareschi, M.; et al. AKT1E17K in human solid tumours. Oncogene 2008, 27, 5648–5650. [Google Scholar] [CrossRef] [Green Version]
- Troxell, M.L. PIK3CA/AKT1 Mutations in Breast Carcinoma: A Comprehensive Review of Experimental and Clinical Studies. J. Clin. Exp. Pathol. 2012, S1-002. [Google Scholar] [CrossRef]
- Shukla, S.; MacLennan, G.T.; Hartman, U.J.; Fu, P.; Resnick, M.I.; Gupta, S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer 2007, 121, 1424–1432. [Google Scholar] [CrossRef]
- Majumder, P.K.; Yeh, J.J.; George, D.J.; Febbo, P.G.; Kum, J.; Xue, Q.; Bikoff, R.; Ma, H.; Kantoff, P.W.; Golub, T.R.; et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: The MPAKT model. Proc. Natl. Acad. Sci. USA 2003, 100, 7841–7846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Sun, A.; Youn, H.; Hong, Y.; Terranova, P.F.; Thrasher, J.; Xu, P.; Spencer, D. Conditional Akt activation promotes androgen-independent progression of prostate cancer. Carcinogenesis 2006, 28, 572–583. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-L.; Xu, P.-Z.; Peng, X.-D.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten± mice. Genome Res. 2006, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study of Akt Blockade with or without Ipatasertib in Abiraterone-Treated Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Maurer, M.; Su, T.; Saal, L.H.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y.; et al. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellwood-Yen, K.; Keilhack, H.; Kunii, K.; Dolinski, B.; Connor, Y.; Hu, K.; Nagashima, K.; O’Hare, E.; Erkul, Y.; Di Bacco, A.; et al. PDK1 Attenuation Fails to Prevent Tumor Formation in PTEN-Deficient Transgenic Mouse Models. Cancer Res. 2011, 71, 3052–3065. [Google Scholar] [CrossRef] [Green Version]
- Magee, J.A.; Chang, L.W.; Stormo, G.D.; Milbrandt, J. Direct, Androgen Receptor-Mediated Regulation of the FKBP5 Gene via a Distal Enhancer Element. Endocrinology 2006, 147, 590–598. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Pratt, C.; Zeeman, M.E.; Schultz, N.; Taylor, B.S.; O’Neill, A.; Castillo-Martin, M.; Nowak, D.G.; Naguib, A.; Grace, D.M.; et al. Identification of PHLPP1 as a Tumor Suppressor Reveals the Role of Feedback Activation in PTEN-Mutant Prostate Cancer Progression. Cancer Cell 2011, 20, 173–186. [Google Scholar] [CrossRef] [Green Version]
- Nowak, D.G.; Katsenelson, K.C.; Watrud, K.E.; Chen, M.; Mathew, G.; D’Andrea, V.D.; Lee, M.F.; Swamynathan, M.M.; Casanova-Salas, I.; Jibilian, M.C.; et al. The PHLPP2 phosphatase is a druggable driver of prostate cancer progression. J. Cell Biol. 2019, 218, 1943–1957. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.; Seshacharyulu, P.; Das, S.; Rachagani, S.; Ponnusamy, M.P.; Yan, Y.; Johansson, S.L.; Datta, K.; Lin, M.F.; Batra, S.K. Impaired expression of protein phosphatase 2A subunits enhances metastatic potential of human prostate cancer cells through activation of AKT pathway. Br. J. Cancer 2013, 108, 2590–2600. [Google Scholar] [CrossRef] [Green Version]
- Malik, N.; Macartney, T.; Hornberger, A.; Anderson, K.E.; Tovell, H.; Prescott, A.R.; Alessi, D.R. Mechanism of activation of SGK3 by growth factors via the Class 1 and Class 3 PI3Ks. Biochem. J. 2018, 475, 117–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basnet, R.; Gong, G.; Li, C.; Wang, M. Serum and glucocorticoid inducible protein kinases (SGKs): A potential target for cancer intervention. Acta Pharm. Sin. B 2018, 8, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Deak, M.; Morrice, N.; Cohen, P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem. J. 1999, 344, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Tessier, M.; Woodgett, J.R. Serum and glucocorticoid-regulated protein kinases: Variations on a theme. J. Cell. Biochem. 2006, 98, 1391–1407. [Google Scholar] [CrossRef] [PubMed]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Chou, M.M.; Hou, W.; Johnson, J.; Graham, L.K.; Lee, M.H.; Chen, C.S.; Newton, A.C.; Schaffhausen, B.S.; Toker, A. Regulation of protein kinase C zeta by PI 3-kinase and PDK-1. Curr. Biol. 1998, 8, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, H.; Nishida, E. The ERK MAP kinase pathway mediates induction of SGK (serum- and glucocorticoid-inducible kinase) by growth factors. Genes Cells 2001, 6, 261–268. [Google Scholar] [CrossRef]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.; Eyers, P.A.; Alessi, D.R. The hVps34- SGK 3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC 1 and tumour growth. EMBO J. 2016, 35, 1902–1922. [Google Scholar] [CrossRef]
- Hayashi, M.; Tapping, R.I.; Chao, T.-H.; Lo, J.-F.; King, C.C.; Yang, Y.; Lee, J.-D. BMK1 Mediates Growth Factor-induced Cell Proliferation through Direct Cellular Activation of Serum and Glucocorticoid-inducible Kinase. J. Biol. Chem. 2001, 276, 8631–8634. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Yamagiwa, Y.; Taffetani, S.; Han, J.; Patel, T. IL-6 activates serum and glucocorticoid kinase via p38α mitogen-activated protein kinase pathway. Am. J. Physiol. Physiol. 2005, 289, C971–C981. [Google Scholar] [CrossRef] [Green Version]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Griend, D.V.; Conzen, S.D.; Szmulewitz, R. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, X.; Liu, Z.; Wang, Y.; Yin, B.; Yu, P.; Duan, X.; Liao, Z.; Chen, Y.; Liu, C.; et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br. J. Cancer 2017, 117, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Gasser, J.A.; Inuzuka, H.; Lau, A.W.; Wei, W.; Beroukhim, R.; Toker, A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol. Cell 2014, 56, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. Second AKT: The rise of SGK in cancer signalling. Growth Factors 2010, 28, 394–408. [Google Scholar] [CrossRef]
- Zhang, Y.; Gan, B.; Liu, D.; Paik, J.-H. FoxO family members in cancer. Cancer Biol. Ther. 2011, 12, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-C.; Jeon, S.-M.; Bhaskar, P.T.; Nogueira, V.; Sundararajan, D.; Tonic, I.; Park, Y.; Hay, N. FoxOs Inhibit mTORC1 and Activate Akt by Inducing the Expression of Sestrin3 and Rictor. Dev. Cell 2010, 18, 592–604. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhou, Y.; Graves, D. FOXO transcription factors: Their clinical significance and regulation. Biomed. Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.-H.; Long, N.P.; Luu, T.-T.-T.; Anh, N.H.; Kwon, S.W.; Lee, S.K. The Dominant Role of Forkhead Box Proteins in Cancer. Int. J. Mol. Sci. 2018, 19, 3279. [Google Scholar] [CrossRef] [Green Version]
- Van Der Heide, L.P.; Hoekman, M.F.M.; Smidt, M.P. The ins and outs of FoxO shuttling: Mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004, 380, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Hyytinen, E.-R.; Saadut, R.; Chen, C.; Paull, L.; Koivisto, P.A.; Vessella, R.L.; Frierson, H.F.; Dong, J.-T. Defining the region(s) of deletion at 6q16-q22 in human prostate cancer. Genes Chromosom. Cancer 2002, 34, 306–312. [Google Scholar] [CrossRef]
- Shukla, S.; Bhaskaran, N.; MacLennan, G.T.; Gupta, S. Deregulation of FoxO3a accelerates prostate cancer progression in TRAMP mice. Prostate 2013, 73, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Blee, A.M.; Wang, D.; An, J.; Pan, Y.; Yan, Y.; Ma, T.; He, Y.; Dugdale, J.; Hou, X.; et al. Loss of FOXO1 Cooperates with TMPRSS2-ERG Overexpression to Promote Prostate Tumorigenesis and Cell Invasion. Cancer Res. 2017, 77, 6524–6537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, B.; Gao, L.; Baranowski, C.; Gillard, B.; Wang, J.; Ransom, R.; Ko, H.-K.; Gelman, I.H. A Genome-Wide RNAi Screen Identifies FOXO4 as a Metastasis-Suppressor through Counteracting PI3K/AKT Signal Pathway in Prostate Cancer. PLoS ONE 2014, 9, e101411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Liang, M.-C.; Ma, J.; Chen, L.; Kozlowski, P.; Qin, W.; Li, D.; Goto, J.; Shimamura, T.; Hayes, D.N.; Meyerson, M.; et al. TSC1 loss synergizes with KRAS activation in lung cancer development in the mouse and confers rapamycin sensitivity. Oncogene 2009, 29, 1588–1597. [Google Scholar] [CrossRef] [Green Version]
- Ho, D.W.H.; Chan, L.K.; Chiu, Y.T.; Xu, I.M.J.; Poon, R.T.P.; Cheung, T.T.; Tang, C.N.; Tang, V.W.L.; Lo, I.L.; Lam, P.W.Y.; et al. TSC1/2mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2016, 66, 1496–1506. [Google Scholar] [CrossRef] [Green Version]
- Kladney, R.D.; Cardiff, R.D.; Kwiatkowski, D.J.; Chiang, G.G.; Weber, J.D.; Arbeit, J.M.; Lu, Z.H. Tuberous sclerosis complex 1: An epithelial tumor suppressor essential to prevent spontaneous prostate cancer in aged mice. Cancer Res. 2010, 70, 8937–8947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Teruya-Feldstein, J.; Behrendt, N.; Chen, Z.; Noda, T.; Hino, O.; Cordon-Cardo, C.; Pandolfi, P.P. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005, 19, 1779–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, N.; Koinuma, J.; Ito, T.; Tsuchiya, E.; Kondo, S.; Nakamura, Y.; Daigo, Y. Activation of an oncogenic TBC1D7 (TBC1 domain family, member 7) protein in pulmonary carcinogenesis. Genes Chromosom. Cancer 2010, 49, 353–367. [Google Scholar] [CrossRef]
- Thien, A.; Prentzell, M.T.; Holzwarth, B.; Kläsener, K.; Kuper, I.; Boehlke, C.; Sonntag, A.G.; Ruf, S.; Maerz, L.; Nitschke, R.; et al. TSC1 Activates TGF-β-Smad2/3 Signaling in Growth Arrest and Epithelial-to-Mesenchymal Transition. Dev. Cell 2015, 32, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Nardella, C.; Chen, Z.; Salmena, L.; Carracedo, A.; Alimonti, A.; Egia, A.; Carver, B.; Gerald, W.; Cordon-Cardo, C.; Pandolfi, P.P. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 2008, 22, 2172–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of mTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway that Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2016, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.R.; Gout, I. Functions and regulation of the 70kDa ribosomal S6 kinases. Int. J. Biochem. Cell Biol. 2011, 43, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Meyuhas, O.; Dreazen, A. Chapter 3 Ribosomal Protein S6 Kinase. Prog. Mol. Biol. Transl. Sci. 2009, 90, 109–153. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, S.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H.; Jhanwar-Uniyal, M. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhou, Y.; Rychahou, P.; Harris, J.W.; Zaytseva, Y.Y.; Liu, J.; Wang, C.; Weiss, H.; Liu, C.; Lee, E.Y.; et al. Deptor is a novel target of Wnt/β-catenin/c-Myc and contributes to colorectal cancer cell growth. Cancer Res. 2018, 78, 3163–3175. [Google Scholar] [CrossRef] [Green Version]
- Catena, V.; Bruno, T.; De Nicola, F.; Goeman, F.; Pallocca, M.; Iezzi, S.; Sorino, C.; Cigliana, G.; Floridi, A.; Blandino, G.; et al. Deptor transcriptionally regulates endoplasmic reticulum homeostasis in multiple myeloma cells. Oncotarget 2016, 7, 70546–70558. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.-H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR Complex 2 Is Required for the Development of Prostate Cancer Induced by Pten Loss in Mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demetriades, C.; Doumpas, N.; Teleman, A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.P.; Frank, A.R.; Jewell, J.L. Amino acid and small GTPase regulation of mTORC1. Cell. Logist. 2017, 7, e1378794. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.-L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Dasgupta, B.; Ju, J.-S.; Sasaki, Y.; Liu, X.; Jung, S.-R.; Higashida, K.; Lindquist, D.; Milbrandt, J. The AMPK β2 Subunit Is Required for Energy Homeostasis during Metabolic Stress. Mol. Cell. Biol. 2012, 32, 2837–2848. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Méry, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Li, C.-F.; Cai, Z.; Zhang, X.; Jin, G.; Zhang, W.-N.; Xu, C.; Wang, C.-Y.; Morrow, J.; Zhang, S.; et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat. Commun. 2018, 9, 4728. [Google Scholar] [CrossRef]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-translational regulation of lipogenesis via AMPK-dependent phosphorylation of insulin-induced gene. Nat. Commun. 2019, 10, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin. Cancer Res. 2015, 21, 3836–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, Y.; Yang, P.Z.; Ahmad, I.; Nixon, C.; Salt, I.P.; Leung, H.Y. AMP-activated protein kinase (AMPK) as a potential therapeutic target independent of PI3K/Akt signaling in prostate cancer. Oncoscience 2014, 1, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2013, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. AMP-activated protein kinase—An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Cespedes, M. The role of LKB1 in lung cancer. Fam. Cancer 2011, 10, 447–453. [Google Scholar] [CrossRef]
- Hindi, S.M.; Sato, S.; Xiong, G.; Bohnert, K.R.; Gibb, A.A.; Gallot, Y.S.; McMillan, J.; Hill, B.G.; Uchida, S.; Kumar, A. TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Penfold, L.; Woods, A.; Muckett, P.; Nikitin, A.Y.; Kent, T.R.; Zhang, S.; Graham, R.; Pollard, A.; Carling, D. CAMKK2 Promotes Prostate Cancer Independently of AMPK via Increased Lipogenesis. Cancer Res. 2018, 78, 6747–6761. [Google Scholar] [CrossRef] [Green Version]
- Gocher, A.M.; Azabdaftari, G.; Euscher, L.M.; Dai, S.; Karacosta, L.G.; Franke, T.F.; Edelman, A.M. Akt activation by Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J. Biol. Chem. 2017, 292, 14188–14204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehenni, H.; Lin-Marq, N.; Buchet-Poyau, K.; Reymond, A.; Collart, M.A.; Picard, D.; Antonarakis, S.E. LKB1 interacts with and phosphorylates PTEN: A functional link between two proteins involved in cancer predisposing syndromes. Hum. Mol. Genet. 2005, 14, 2209–2219. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Clarke, A. Energy sensing and cancer: LKB1 function and lessons learnt from Peutz–Jeghers syndrome. Semin. Cell Dev. Biol. 2016, 52, 21–29. [Google Scholar] [CrossRef]
- Pearson, H.; McCarthy, A.; Collins, C.M.; Ashworth, A.; Clarke, A. Lkb1 Deficiency Causes Prostate Neoplasia in the Mouse. Cancer Res. 2008, 68, 2223–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanova, I.; Zúñiga-García, P.; Caro-Maldonado, A.; Fernandez-Ruiz, S.; Salvador, F.; Martín-Martín, N.; Zabala-Letona, A.; Nuñez-Olle, M.; Torrano, V.; Camacho, L.; et al. Genetic manipulation of LKB1 elicits lethal metastatic prostate cancer. J. Exp. Med. 2020, 217, e20191787. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Cai, F.; Liu, X.; Guo, L. LKB1 suppresses proliferation and invasion of prostate cancer through hedgehog signaling pathway. Int. J. Clin. Exp. Pathol. 2014, 7, 8480–8488. [Google Scholar]
- Budanov, A.; Karin, M. p53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2014, 29, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Xu, Y.; Liu, J.; Ye, J.; Yuan, W.; Jiang, H.; Wang, Z.; Jiang, H.; Wan, J. Recent Insights into the Biological Functions of Sestrins in Health and Disease. Cell. Physiol. Biochem. 2017, 43, 1731–1741. [Google Scholar] [CrossRef]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.-L.; Karin, M.; Budanov, A. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [Green Version]
- Cordani, M.; Sanchez-Alvarez, M.; Strippoli, R.; Bazhin, A.; Donadelli, M. Sestrins at the Interface of ROS Control and Autophagy Regulation in Health and Disease. Oxidative Med. Cell. Longev. 2019, 2019, 1283075. [Google Scholar] [CrossRef] [PubMed]
- Pasha, M.; Eid, A.; Eid, A.A.; Gorin, Y.; Munusamy, S. Sestrin2 as a Novel Biomarker and Therapeutic Target for Various Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jones, S.J.M.; Marra, M.A.; Sadar, M.D. Identification of genes targeted by the androgen and PKA signaling pathways in prostate cancer cells. Oncogene 2006, 25, 7311–7323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaly, S.R.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.-S.; Tsai, W.-L.; Liu, P.-F.; Goan, Y.-G.; Lin, C.-W.; Tseng, H.-H.; Lee, C.-H.; Shu, C.-W. The MAP3K7-mTOR Axis Promotes the Proliferation and Malignancy of Hepatocellular Carcinoma Cells. Front. Oncol. 2019, 9, 474. [Google Scholar] [CrossRef]
- Kluth, M.; Hesse, J.; Heinl, A.; Krohn, A.; Steurer, S.; Sirma, H.; Simon, R.; Mayer, P.-S.; Schumacher, U.; Grupp, K.; et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod. Pathol. 2013, 26, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Shi, L.; Cimic, A.; Romero, L.; Sui, G.; Lees, C.J.; Cline, J.M.; Seals, D.F.; Sirintrapun, J.S.; McCoy, T.P.; et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res. 2012, 72, 2833–2843. [Google Scholar] [CrossRef] [Green Version]
- Bosman, M.C.J.; Schepers, H.; Jaques, J.; Brouwers-Vos, A.Z.; Quax, W.J.; Schuringa, J.J.; Vellenga, E. The TAK1-NF-κB axis as therapeutic target for AML. Blood 2014, 124, 3130–3140. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Sathe, A.; Chalaud, G.; Oppolzer, I.; Wong, K.Y.; Von Busch, M.; Schmid, S.C.; Tong, Z.; Retz, M.; Gschwend, J.E.; Schulz, W.A.; et al. Parallel PI3K, AKT and mTOR inhibition is required to control feedback loops that limit tumor therapy. PLoS ONE 2018, 13, e0190854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozengurt, E.; Soares, H.P.; Sinnet-Smith, J. Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: An unintended consequence leading to drug resistance. Mol. Cancer Ther. 2014, 13, 2477–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Lipovsky, A.I.; Dibble, C.; Sahin, M.; Manning, B.D. S6K1 Regulates GSK3 under Conditions of mTOR-Dependent Feedback Inhibition of Akt. Mol. Cell 2006, 24, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2006, 26, 1932–1940. [Google Scholar] [CrossRef] [Green Version]
- Margolis, B.; Skolnik, E.Y. Activation of Ras by receptor tyrosine kinases. J. Am. Soc. Nephrol. 1994, 5, 1288–1299. [Google Scholar]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Medarde, A.; Santos, E. Ras in Cancer and Developmental Diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, C.R.; Long, J.; Laidler, P.; Mijatović, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [Green Version]
- Copps, K.D.; White, M. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.; Asara, J.M.; Manning, B.D. Characterization of Rictor Phosphorylation Sites Reveals Direct Regulation of mTOR Complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suire, S.; Hawkins, P.; Stephens, L. Activation of phosphoinositide 3-kinase gamma by Ras. Curr. Biol. 2002, 12, 1068–1075. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Carrière, A.; Cargnello, M.; Julien, L.-A.; Gao, H.; Bonneil, É.; Thibault, P.; Roux, P.P. Oncogenic MAPK Signaling Stimulates mTORC1 Activity by Promoting RSK-Mediated Raptor Phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef] [Green Version]
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 Phosphorylate Raptor to Promote Ras-dependent Activation of mTOR Complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Lara, R.; Seckl, M.J.; Pardo, O.E. The p90 RSK Family Members: Common Functions and Isoform Specificity. Cancer Res. 2013, 73, 5301–5308. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, S. Phosphorylation and Regulation of Raf by Akt (Protein Kinase B). Science 1999, 286, 1741–1744. [Google Scholar] [CrossRef]
- Guan, K.-L. Negative regulation of the serine/threonine kinase B-Raf by Akt. J. Biol. Chem. 2000, 275, 275. [Google Scholar] [CrossRef]
- Lone, M.-U.-D.; Miyan, J.; Asif, M.; Malik, S.A.; Dubey, P.; Singh, V.; Singh, K.; Mitra, K.; Pandey, D.; Haq, W.; et al. Direct physical interaction of active Ras with mSIN1 regulates mTORC2 signaling. BMC Cancer 2019, 19, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Birle, D.C.; Tannock, I.F. Effects of the Mammalian Target of Rapamycin Inhibitor CCI-779 Used Alone or with Chemotherapy on Human Prostate Cancer Cells and Xenografts. Cancer Res. 2005, 65, 2825–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, R.J.; Jac, J.; Mohammad, T.; Saxena, S. Pilot Study of Rapamycin in Patients with Hormone-Refractory Prostate Cancer. Clin. Genitourin. Cancer 2008, 6, 97–102. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Kinkade, C.W.; Castillo-Martin, M.; Puzio-Kuter, A.; Yan, J.; Foster, T.H.; Gao, H.; Sun, Y.; Ouyang, X.; Gerald, W.L.; Cordon-Cardo, C.; et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J. Clin. Investig. 2008, 118, 3051–3064. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.E.; Marlein, C.; Walker, H.F.; Frame, F.M.; Mann, V.M.; Simms, M.S.; Davies, B.R.; Collins, A.T.; Maitland, N.J. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017, 8, 56698–56713. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yan, H.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, F.; Gagnon, A.; Veilleux, A.; Sorisky, A.; Marette, A. Activation of the Mammalian Target of Rapamycin Pathway Acutely Inhibits Insulin Signaling to Akt and Glucose Transport in 3T3-L1 and Human Adipocytes. Endocrinology 2005, 146, 1328–1337. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Yoon, S.-O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villén, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate That Negatively Regulates Insulin Signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Outmezguine, V.R.-; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biggs, W.H.; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. USA 2011, 109, 2718–2723. [Google Scholar] [CrossRef] [Green Version]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.A.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzmán, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2547–2557. [Google Scholar] [CrossRef]
- Sommer, E.M.; Dry, H.; Cross, D.; Guichard, S.; Davies, B.R.; Alessi, D.R. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem. J. 2013, 452, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Kobayashi, T.; Floc’H, N.; Kinkade, C.W.; Aytes, A.; Dankort, D.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; McMahon, M.; et al. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer Res. 2012, 72, 4765–4776. [Google Scholar] [CrossRef] [Green Version]
- Jefferies, M.T.; Cox, A.C.; Shorning, B.Y.; Meniel, V.; Griffiths, D.; Kynaston, H.G.; Smalley, M.; Clarke, A.R. PTEN loss and activation of K-RAS and β-catenin cooperate to accelerate prostate tumourigenesis. J. Pathol. 2017, 243, 442–456. [Google Scholar] [CrossRef]
- Toren, P.; Kim, S.; Johnson, F.; Zoubeidi, A. Combined AKT and MEK Pathway Blockade in Pre-Clinical Models of Enzalutamide-Resistant Prostate Cancer. PLoS ONE 2016, 11, e0152861. [Google Scholar] [CrossRef]
- Turke, A.B.; Song, Y.; Costa, C.; Cook, R.; Arteaga, C.L.; Asara, J.M.; Engelman, J.A. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012, 72, 3228–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gioeli, D.; Wunderlich, W.; Sebolt-Leopold, J.; Bekiranov, S.; Wulfkuhle, J.D.; Petricoin, E.F.; Conaway, M.; Weber, M.J. Compensatory pathways induced by MEK inhibition are effective drug targets for combination therapy against castration-resistant prostate cancer. Mol. Cancer Ther. 2011, 10, 1581–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabb, S.J.; Birtle, A.J.; Martin, K.; Downs, N.; Ratcliffe, I.; Maishman, T.; Ellis, M.; Griffiths, G.; Thompson, S.; Ksiazek, L.; et al. ProCAID: A phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Investig. New Drugs 2017, 35, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Gillessen, S.; Gilson, C.; James, N.D.; Adler, A.; Sydes, M.R.; Clarke, N. Repurposing Metformin as Therapy for Prostate Cancer within the STAMPEDE Trial Platform. Eur. Urol. 2016, 70, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Guigas, B.; Garcia, N.S.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2011, 122, 253–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, H.P.; Ni, Y.; Kisfalvi, K.; Sinnett-Smith, J.; Rozengurt, E. Different Patterns of Akt and ERK Feedback Activation in Response to Rapamycin, Active-Site mTOR Inhibitors and Metformin in Pancreatic Cancer Cells. PLoS ONE 2013, 8, e57289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokinen, E.; Koivunen, J. MEK and PI3K inhibition in solid tumors: Rationale and evidence to date. Ther. Adv. Med. Oncol. 2015, 7, 170–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Gounder, M.; Rodon, J.; Janku, F.; Lolkema, M.P.; Stephenson, J.J.; Bedard, P.L.; Schuler, M.; Sessa, C.; Lorusso, P.; et al. Phase Ib Study of Combination Therapy with MEK Inhibitor Binimetinib and Phosphatidylinositol 3-Kinase Inhibitor Buparlisib in Patients with Advanced Solid Tumors with RAS/RAF Alterations. Oncologist 2019, 25, e160–e169. [Google Scholar] [CrossRef] [Green Version]
- Tindall, N.J.; Lonergan, P.E. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [CrossRef]
- Zhou, Y.; Bolton, E.C.; Jones, J.O. Androgens and androgen receptor signaling in prostate tumorigenesis. J. Mol. Endocrinol. 2014, 54, R15–R29. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2014, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2010, 32, 81–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Balk, S.P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 2011, 18, R175–R182. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Chung, Y.-M.; Kovac, E.; Zhu, Z.; Li, J.; Magi-Galluzzi, C.; Stephenson, A.J.; Klein, E.A.; Sharifi, N. Direct Metabolic Interrogation of Dihydrotestosterone Biosynthesis from Adrenal Precursors in Primary Prostatectomy Tissues. Clin. Cancer Res. 2017, 23, 6351–6362. [Google Scholar] [CrossRef] [Green Version]
- Dehm, S.M.; Tindall, N.J. Androgen Receptor Structural and Functional Elements: Role and Regulation in Prostate Cancer. Mol. Endocrinol. 2007, 21, 2855–2863. [Google Scholar] [CrossRef]
- Lamont, K.R.; Tindall, N.J. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol. Endocrinol. 2011, 25, 897–907. [Google Scholar] [CrossRef]
- Zamagni, A.; Cortesi, M.; Zanoni, M.; Tesei, A. Non-nuclear AR Signaling in Prostate Cancer. Front. Chem. 2019, 7, 651. [Google Scholar] [CrossRef]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef]
- Saranyutanon, S.; Srivastava, S.K.; Pai, S.; Singh, S.; Singh, A.P. Therapies Targeted to Androgen Receptor Signaling Axis in Prostate Cancer: Progress, Challenges, and Hope. Cancers 2019, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Murillo, H.; Huang, H.; Schmidt, L.J.; Smith, D.I.; Tindall, N.J. Role of PI3K Signaling in Survival and Progression of LNCaP Prostate Cancer Cells to the Androgen Refractory State. Endocrinology 2001, 142, 4795–4805. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Gleave, M. Co-targeting driver pathways in prostate cancer: Two birds with one stone. EMBO Mol. Med. 2018, 10, e8928. [Google Scholar] [CrossRef] [PubMed]
- Audet-Walsh, É.; Dufour, C.R.; Yee, T.; Zouanat, F.Z.; Yan, M.; Kalloghlian, G.; Vernier, M.; Caron, M.; Bourque, G.; Scarlata, E.; et al. Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer. Genes Dev. 2017, 31, 1228–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Morales, C.; Cooke, L.S.; Johnson, B.; Somer, B.; Mahadevan, D. Reciprocal feedback inhibition of the androgen receptor and PI3K as a novel therapy for castrate-sensitive and -resistant prostate cancer. Oncotarget 2015, 6, 41976–41987. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.R.; Beraldi, E.; Fazli, L.; Kim, S.; Thaper, D.; Gleave, M.E.; Zoubeidi, A. Synergistic Targeting of PI3K/AKT Pathway and Androgen Receptor Axis Significantly Delays Castration-Resistant Prostate Cancer Progression In Vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, E.; Zhou, W.; Lee-Hoeflich, S.T.; Truong, T.; Haverty, P.M.; Eastham-Anderson, J.; Lewin-Koh, N.; Günter, B.; Belvin, M.; Murray, L.J.; et al. Suppression of HER2/HER3-Mediated Growth of Breast Cancer Cells with Combinations of GDC-0941 PI3K Inhibitor, Trastuzumab, and Pertuzumab. Clin. Cancer Res. 2009, 15, 4147–4156. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, N.P.; Liu, Y.; Majumder, S.; Warren, M.R.; Parker, C.E.; Mohler, J.L.; Earp, H.S.; Whang, Y.E. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl. Acad. Sci. USA 2007, 104, 8438–8443. [Google Scholar] [CrossRef] [Green Version]
- Mellinghoff, I.K.; Vivanco, I.; Kwon, A.; Tran, C.; Wongvipat, J.; Sawyers, C.L. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 2004, 6, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Yeh, S.; Lin, H.-K.; Kang, H.-Y.; Thin, T.H.; Chang, C. From HER2/Neu signal cascade to androgen receptor and its coactivators: A novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc. Natl. Acad. Sci. USA 1999, 96, 5458–5463. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Li, S.; Gan, L.; Kao, T.P.; Huang, H. A Transcription-Independent Function of FOXO1 in Inhibition of Androgen-Independent Activation of the Androgen Receptor in Prostate Cancer Cells. Cancer Res. 2008, 68, 10290–10299. [Google Scholar] [CrossRef] [Green Version]
- Bowen, C.; Ostrowski, M.C.; Leone, G.; Gelmann, E.P. Loss of PTEN Accelerates NKX3.1 Degradation to Promote Prostate Cancer Progression. Cancer Res. 2019, 79, 4124–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, P.Y.; Chang, C.W.; Chng, K.R.; Wansa, K.D.S.A.; Sung, W.-K.; Cheung, E. Integration of Regulatory Networks by NKX3-1 Promotes Androgen-Dependent Prostate Cancer Survival. Mol. Cell. Biol. 2011, 32, 399–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.-Y.; Jiao, J.; Xin, L.; Chang, C.-J.; Wang, S.; Gao, J.; Gleave, M.E.; Witte, O.N.; Liu, X.; Wu, H. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell 2006, 9, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Bailey, C.; Ng, C.; Tiffen, J.; Thoeng, A.; Minhas, V.; Lehman, M.L.; Hendy, S.C.; Buchanan, G.; Nelson, C.C.; et al. Androgen Receptor and Nutrient Signaling Pathways Coordinate the Demand for Increased Amino Acid Transport during Prostate Cancer Progression. Cancer Res. 2011, 71, 7525–7536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Hu, M.C.; Makino, K.; Spohn, B.; Bartholomeusz, G.; Yan, D.H.; Hung, M.C. HER-2/neu promotes androgen-independent survival and growth of prostate cancer cells through the Akt pathway. Cancer Res. 2000, 60, 6841–6845. [Google Scholar] [PubMed]
- Lin, H.-K.; Yeh, S.; Kang, H.-Y.; Chang, C. Akt suppresses androgen-induced apoptosis by phosphorylating and inhibiting androgen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 7200–7205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattner, M.; Liu, D.; Robinson, B.D.; Huang, D.; Poliakov, A.; Gao, D.; Nataraj, S.; Deonarine, L.D.; Augello, M.A.; Sailer, V.; et al. SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Mani, R.S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today 2014, 19, 1498–1502. [Google Scholar] [CrossRef] [Green Version]
- Agoulnik, I.U.; Weigel, N.L. Coactivator selective regulation of androgen receptor activity. Steroids 2009, 74, 669–674. [Google Scholar] [CrossRef] [Green Version]
- Ferry, C.; Gaouar, S.; Fischer, B.; Boeglin, M.; Paul, N.; Samarut, E.; Piskunov, A.; Pankotai-Bodó, G.; Brino, L.; Rochette-Egly, C. Cullin 3 mediates SRC-3 ubiquitination and degradation to control the retinoic acid response. Proc. Natl. Acad. Sci. USA 2011, 108, 20603–20608. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Wang, C.; Deng, Y.; Yu, L.; Huang, H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. 2014, 6, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, S.; Manin, M.; Beaudoin, C.; Leotoing, L.; Communal, Y.; Veyssiere, G.; Morel, L. Androgen Receptor Mediates Non-genomic Activation of Phosphatidylinositol 3-OH Kinase in Androgen-sensitive Epithelial Cells. J. Biol. Chem. 2003, 279, 14579–14586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolinsky, M.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Chi, K.N.; Castellano, D.; De Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.-P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.X.; Hsieh, A.C.; Kim, W.; Friedlander, T.; Lin, A.M.; Louttit, M.; Ryan, C.J. A Phase I Study of Abiraterone Acetate Combined with BEZ235, a Dual PI3K/mTOR Inhibitor, in Metastatic Castration Resistant Prostate Cancer. Oncologist 2017, 22, 503-e43. [Google Scholar] [CrossRef] [Green Version]
- D’Abronzo, L.S.; Bose, S.; Crapuchettes, M.E.; Beggs, R.E.; Vinall, R.L.; Tepper, C.G.; Siddiqui, S.; Mudryj, M.; Melgoza, F.U.; Durbin-Johnson, B.P.; et al. The androgen receptor is a negative regulator of eIF4E phosphorylation at S209: Implications for the use of mTOR inhibitors in advanced prostate cancer. Oncogene 2017, 36, 6359–6373. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Loh, K.M.; Nusse, R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Murillo-Garzón, V.; Kypta, R. WNT signalling in prostate cancer. Nat. Rev. Urol. 2017, 14, 683–696. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2012, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Logan, S.K. Revisiting the role of Wnt/β-catenin signaling in prostate cancer. Mol. Cell. Endocrinol. 2017, 462, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Sansom, O.J. Role of Wnt signalling in advanced prostate cancer. J. Pathol. 2018, 245, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Lanman, N.A.; Pascuzzi, P.; Gupta, S.; Liu, X. Inhibition of the Wnt/β-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velho, P.I.; Fu, W.; Wang, H.; Mirkheshti, N.; Qazi, F.; Lima, F.A.; Shaukat, F.; Carducci, M.A.; Denmeade, S.R.; Paller, C.J.; et al. Wnt-pathway Activating Mutations Are Associated with Resistance to First-line Abiraterone and Enzalutamide in Castration-resistant Prostate Cancer. Eur. Urol. 2019, 77, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Wickström, M.; Baryawno, N. Wingless/β-catenin signaling as a modulator of chemoresistance in cancer. Mol. Cell. Oncol. 2016, 3, e1131356. [Google Scholar] [CrossRef] [Green Version]
- Rajan, P.; Sudbery, I.; Villasevil, M.E.M.; Mui, E.; Fleming, J.; Davis, M.; Ahmad, I.; Edwards, J.; Sansom, O.J.; Sims, D.; et al. Next-generation Sequencing of Advanced Prostate Cancer Treated with Androgen-deprivation Therapy. Eur. Urol. 2014, 66, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Song, L.-N.; Gelmann, E.P. Interaction of -Catenin and TIF2/GRIP1 in Transcriptional Activation by the Androgen Receptor. J. Biol. Chem. 2005, 280, 37853–37867. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chen, M.-W.; Terry, S.; Vacherot, F.; Bemis, D.L.; Capodice, J.; Kitajewski, J.; De La Taille, A.; Benson, M.C.; Guo, Y.; et al. Complex regulation of human androgen receptor expression by Wnt signaling in prostate cancer cells. Oncogene 2006, 25, 3436–3444. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; Sadar, M.D. Crosstalk between the androgen receptor and beta-catenin in castrate-resistant prostate cancer. Cancer Res. 2008, 68, 9918–9927. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Brzezinska, E.A.; Repiscak, P.; Ahmad, I.; Mui, E.; Gao, M.; Blomme, A.; Harle, V.; Tan, E.H.; Malviya, G.; et al. Activation of β-Catenin Cooperates with Loss of Pten to Drive AR-Independent Castration-Resistant Prostate Cancer. Cancer Res. 2019, 80, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.; Phesse, T.J.; Clarke, A. K-ras and Wnt Signaling Synergize to Accelerate Prostate Tumorigenesis in the Mouse. Cancer Res. 2009, 69, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruxvoort, K.J.; Charbonneau, H.M.; Giambernardi, T.A.; Goolsby, J.C.; Qian, C.-N.; Zylstra, C.R.; Robinson, D.R.; Roy-Burman, P.; Shaw, A.; Buckner-Berghuis, B.D.; et al. Inactivation of Apc in the Mouse Prostate Causes Prostate Carcinoma. Cancer Res. 2007, 67, 2490–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenbaum, S.P.; Ordóñez-Morán, P.; Puig, I.; Chicote, I.; Arqués, O.; Landolfi, S.; Fernández, Y.; Camacho, J.R.H.; Gispert, J.D.; Mendizabal, L.; et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat. Med. 2012, 18, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persad, A.; Venkateswaran, G.; Hao, L.; Garcia, M.E.; Yoon, J.; Sidhu, J.; Persad, S. Active β-catenin is regulated by the PTEN/PI3 kinase pathway: A role for protein phosphatase PP2A. Genes Cancer 2017, 7, 368–382. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yin, J.; Wang, H.; Jiang, G.; Deng, M.; Zhang, G.; Bu, X.; Cai, S.; Du, J.; He, Z. FOXO3a modulates WNT/β-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells. Cell. Signal. 2015, 27, 510–518. [Google Scholar] [CrossRef]
- Sinner, D.; Kordich, J.J.; Spence, J.R.; Opoka, R.; Rankin, S.; Lin, S.-C.J.; Jonatan, D.; Zorn, A.M.; Wells, J.M. Sox17 and Sox4 Differentially Regulate β-Catenin/T-Cell Factor Activity and Proliferation of Colon Carcinoma Cells. Mol. Cell. Biol. 2007, 27, 7802–7815. [Google Scholar] [CrossRef] [Green Version]
- Bilir, B.; Osunkoya, A.O.; Wiles, W.G.; Sannigrahi, S.; Lefebvre, V.; Metzger, D.; Spyropoulos, D.D.; Martin, W.D.; Moreno, C.S. SOX4 Is Essential for Prostate Tumorigenesis Initiated by PTEN Ablation. Cancer Res. 2015, 76, 1112–1121. [Google Scholar] [CrossRef] [Green Version]
- Conde-Perez, A.; Gros, G.; Longvert, C.; Pedersen, M.; Petit, V.; Aktary, Z.; Viros, A.; Gesbert, F.; Delmas, V.; Rambow, F.; et al. A caveolin-dependent and PI3K/AKT-independent role of PTEN in β-catenin transcriptional activity. Nat. Commun. 2015, 6, 8093. [Google Scholar] [CrossRef]
- Wu, D.; Pan, W. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Buller, C.L.; Loberg, R.D.; Fan, M.-H.; Zhu, Q.; Park, J.L.; Vesely, E.; Inoki, K.; Guan, K.-L.; Brosius, F.C. A GSK-3/TSC2/mTOR pathway regulates glucose uptake and GLUT1 glucose transporter expression. Am. J. Physiol. Physiol. 2008, 295, C836–C843. [Google Scholar] [CrossRef] [Green Version]
- Evangelisti, C.; Chiarini, F.; Paganelli, F.; Marmiroli, S.; Martelli, A.M. Crosstalks of GSK3 signaling with the mTOR network and effects on targeted therapy of cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, V.W.; Chen, R.-H.; McCormick, F. Differential Regulation of Glycogen Synthase Kinase 3β by Insulin and Wnt Signaling. J. Biol. Chem. 2000, 275, 32475–32481. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.S.; Mahmoudi, T.; Danenberg, E.; Bejaoui, I.; De Lau, W.; Korswagen, H.C.; Schutte, M.; Clevers, H. Phosphatidylinositol 3-Kinase Signaling Does Not Activate the Wnt Cascade. J. Biol. Chem. 2009, 284, 35308–35313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravitz, M.J.; Chen, L.; Lynch, M.; Schmidt, E.V. c-myc Repression of TSC2 contributes to control of translation initiation and Myc-induced transformation. Cancer Res. 2007, 67, 11209–11217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csibi, A.; Blenis, J. Hippo–YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012, 14, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S.; Information, P.E.K.F.C. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Aznar, N.; Midde, K.K.; Dunkel, Y.; Lopez-Sanchez, I.; Pavlova, Y.; Marivin, A.; Barbazan, J.; Murray, F.; Nitsche, U.; Janssen, K.-P.; et al. Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. eLife 2015, 4, e07091. [Google Scholar] [CrossRef]
- Hojjat-Farsangi, M.; Moshfegh, A.; Daneshmanesh, A.H.; Khan, S.; Mikaelsson, E.; Österborg, A.; Mellstedt, H. The receptor tyrosine kinase ROR1—An oncofetal antigen for targeted cancer therapy. Semin. Cancer Biol. 2014, 29, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Gujral, T.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, E.; Pessin, J.E.; Kurland, I.J.; Schwartz, G.J.; Bastie, C.C. Fyn-Dependent Regulation of Energy Expenditure and Body Weight Is Mediated by Tyrosine Phosphorylation of LKB1. Cell Metab. 2010, 11, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.; Wilkins, J.A.; Reed, K.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Marderosian, M.; Sharma, A.; Funk, A.P.; Vartanian, R.; Masri, J.; Jo, O.D.; Gera, J. Tristetraprolin regulates Cyclin D1 and c-Myc mRNA stability in response to rapamycin in an Akt-dependent manner via p38 MAPK signaling. Oncogene 2006, 25, 6277–6290. [Google Scholar] [CrossRef] [Green Version]
- Gera, J.; Mellinghoff, I.K.; Shi, Y.; Rettig, M.B.; Tran, C.; Hsu, J.-H.; Sawyers, C.L.; Lichtenstein, A. AKT Activity Determines Sensitivity to Mammalian Target of Rapamycin (mTOR) Inhibitors by Regulating Cyclin D1 and c-mycExpression. J. Biol. Chem. 2003, 279, 2737–2746. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Sharma, A.; Wu, H.; Lichtenstein, A.; Gera, J. Cyclin D1 and c-myc Internal Ribosome Entry Site (IRES)-dependent Translation Is Regulated by AKT Activity and Enhanced by Rapamycin through a p38 MAPK- and ERK-dependent Pathway. J. Biol. Chem. 2005, 280, 10964–10973. [Google Scholar] [CrossRef] [Green Version]
- Wall, M.; Poortinga, G.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D.; McArthur, G. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood 2008, 112, 2305–2317. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by Glycogen Synthase Kinase-3 Controls c-Myc Proteolysis and Subnuclear Localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [Green Version]
- Griend, D.V.; Litvinov, I.V.; Isaacs, J.T. Conversion of Androgen Receptor Signaling From a Growth Suppressor in Normal Prostate Epithelial Cells to an Oncogene in Prostate Cancer Cells Involves a Gain of Function in c-Myc Regulation. Int. J. Biol. Sci. 2014, 10, 627–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, L.; Van Der Schoor, F.; Dalrymple, S.L.; Isaacs, J.T. Androgen receptor (AR) suppresses normal human prostate epithelial cell proliferation via AR/β-catenin/TCF-4 complex inhibition ofc-MYCtranscription. Prostate 2014, 74, 1118–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barfeld, S.J.; Urbanucci, A.; Itkonen, H.M.; Fazli, L.; Hicks, J.L.; Thiede, B.; Rennie, P.S.; Yegnasubramanian, S.; DeMarzo, A.M.; Mills, I.G. c-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks. EBioMed. 2017, 18, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.J.; Pearson, R.B.; Hannan, R.D.; Furic, L. Therapeutic Approaches Targeting MYC-Driven Prostate Cancer. Genes 2017, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Ye, H.; He, H.H.; Gerrin, S.J.; Chen, S.; Tanenbaum, B.A.; Cai, C.; Sowalsky, A.G.; He, L.; Wang, H.; et al. SOX9 drives WNT pathway activation in prostate cancer. J. Clin. Investig. 2016, 126, 1745–1758. [Google Scholar] [CrossRef] [Green Version]





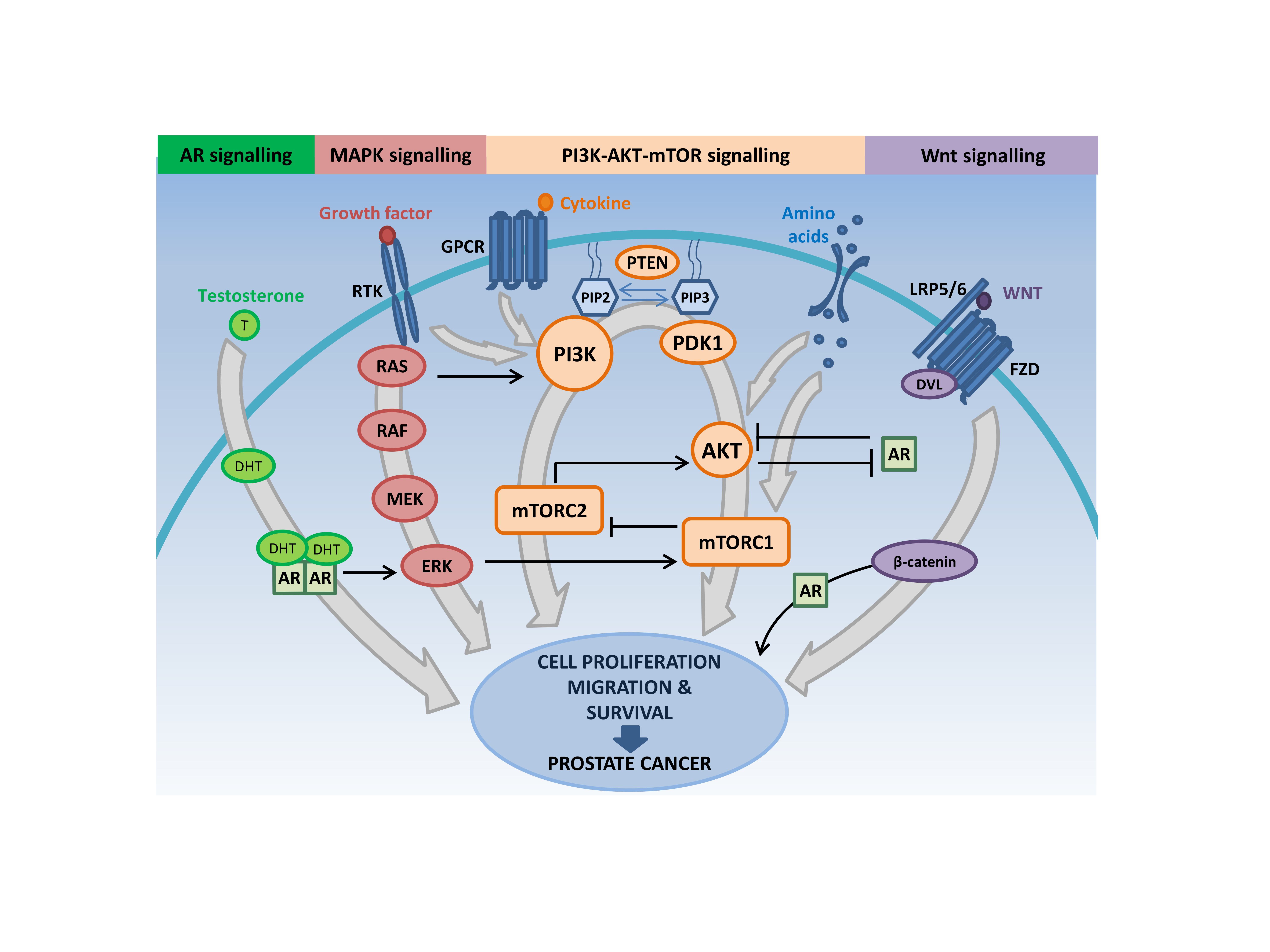{kind=link}
{kind=link}
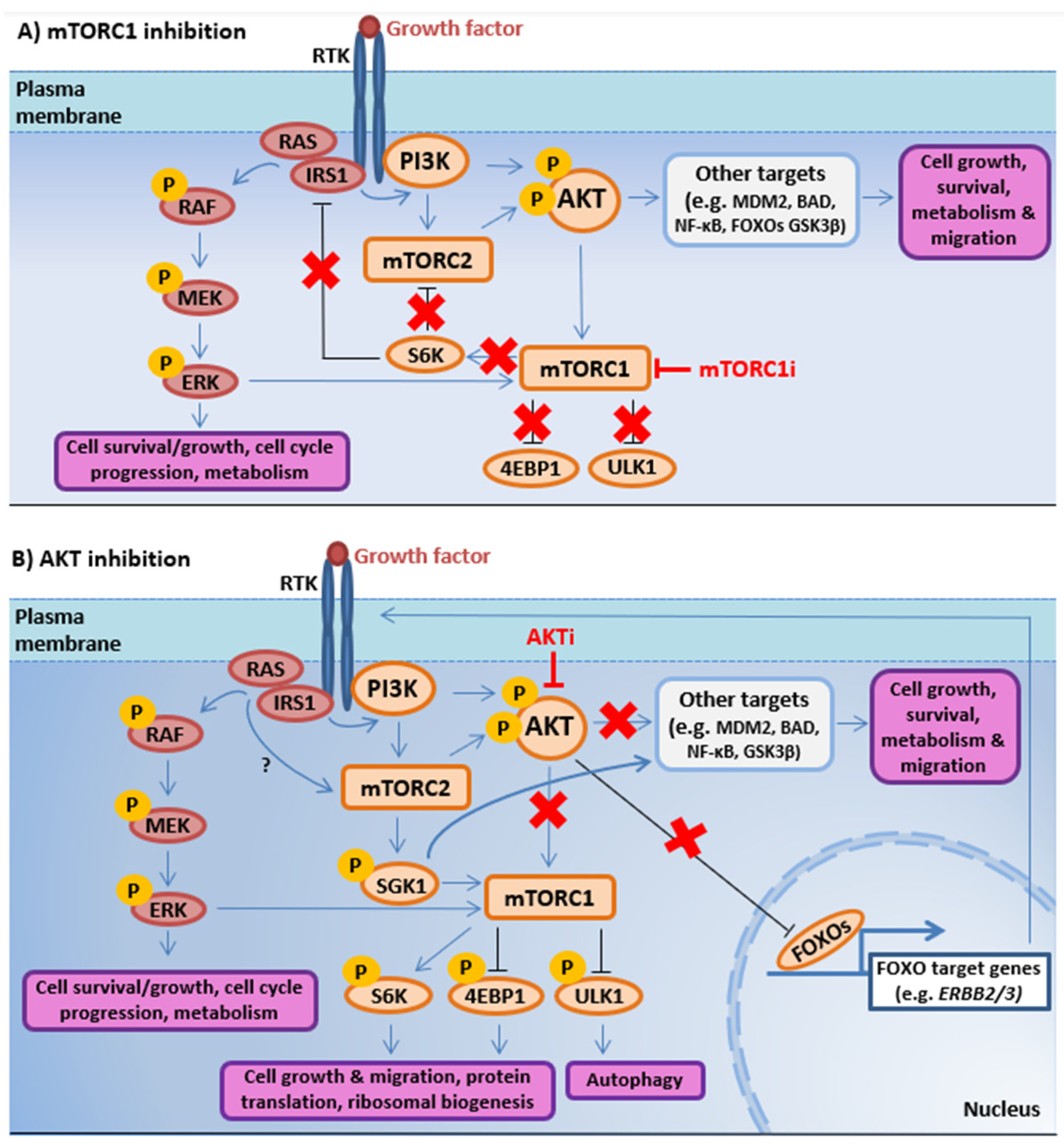{kind=link}
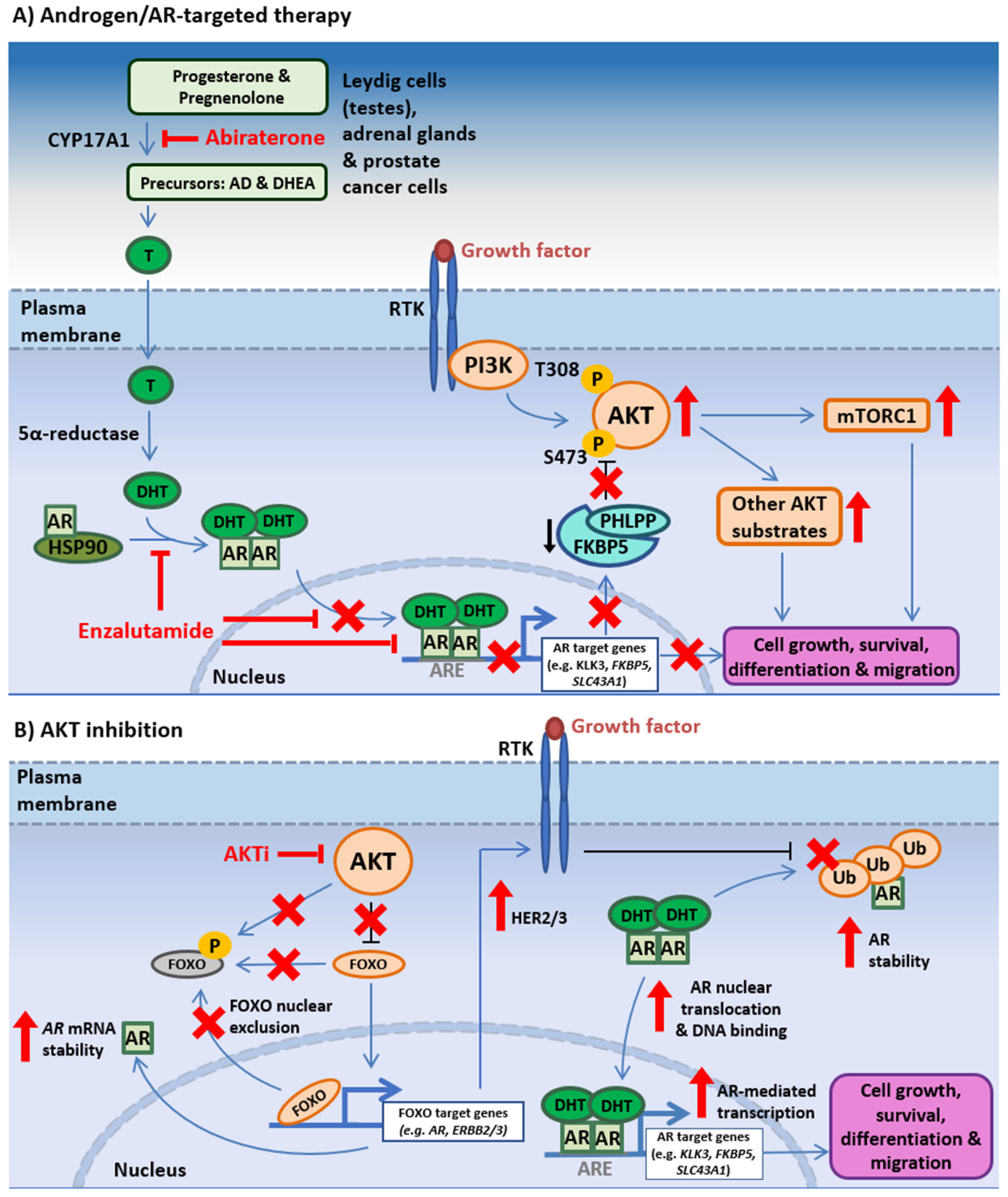{kind=link}
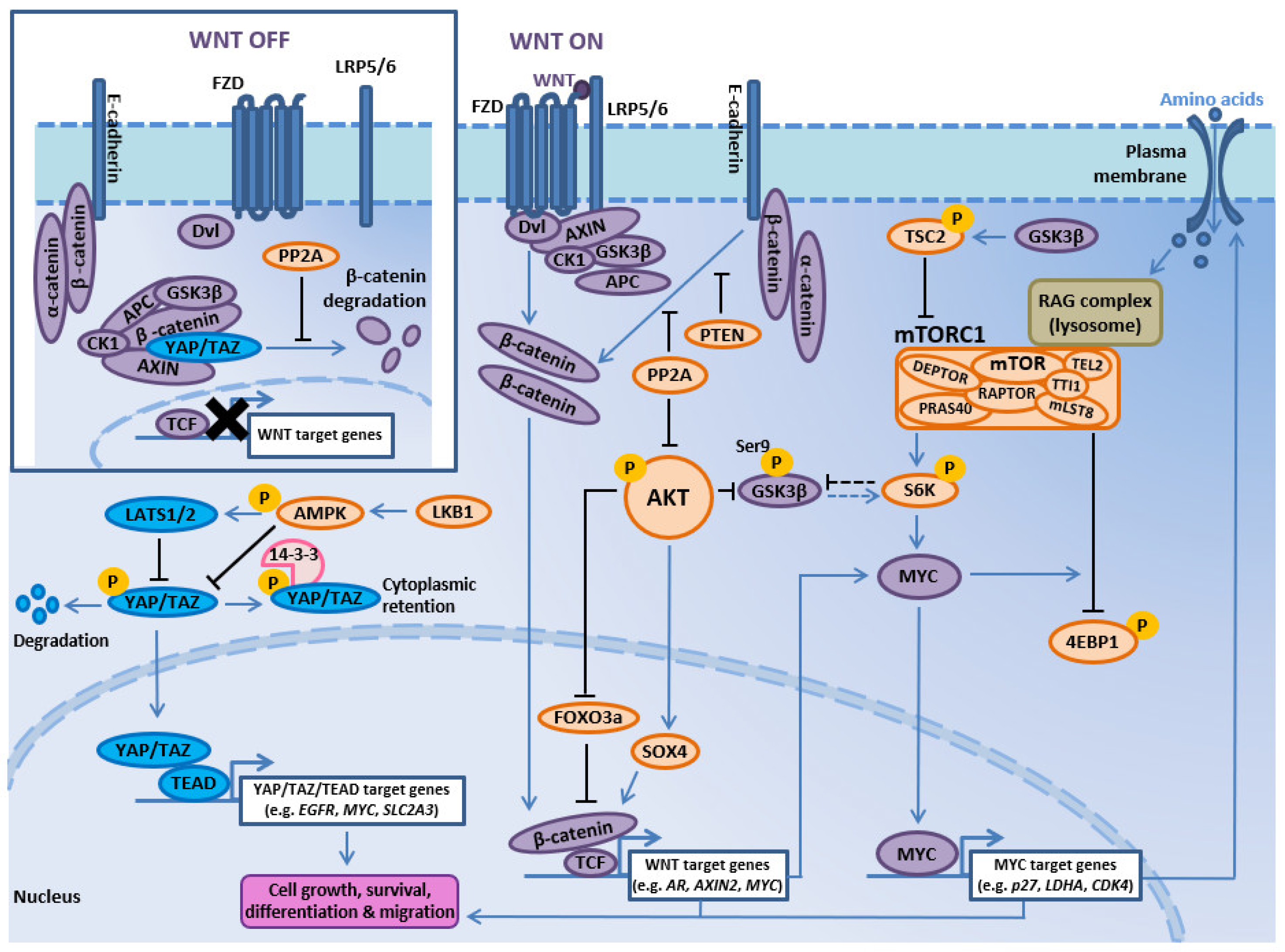{kind=link}
| Common Types of Genetic Alterations in PI3K-AKT-mTOR Pathway Genes | Frequency in Prostate Cancer 1 |
|---|---|
| PTEN deletion/mutation | 16.4–32.0% |
| DEPTOR amplification | 5.1–21.4% |
| SGK mutation/amplification | 5.6–20.5% (SGK3) |
| 0.2–2.7% (SGK1) | |
| FOXO deletion | 0.0–15.2% (FOXO1) |
| 4.5–13.4% (FOXO3) | |
| MAP3K7 deletion | 5.9–14.8% |
| RRAGD deletion | 6.5–14.4% |
| SESN1 mutation/deletion | 5.4–13.6% |
| PIK3CA mutation/amplification | 5.5–11.5% |
| PIK3C2B mutation/amplification | 1.4–11.5% |
| PDPK1 amplification | 0–8.1% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. https://doi.org/10.3390/ijms21124507
Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. International Journal of Molecular Sciences. 2020; 21(12):4507. https://doi.org/10.3390/ijms21124507
Chicago/Turabian StyleShorning, Boris Y., Manisha S. Dass, Matthew J. Smalley, and Helen B. Pearson. 2020. "The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling" International Journal of Molecular Sciences 21, no. 12: 4507. https://doi.org/10.3390/ijms21124507
APA StyleShorning, B. Y., Dass, M. S., Smalley, M. J., & Pearson, H. B. (2020). The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. International Journal of Molecular Sciences, 21(12), 4507. https://doi.org/10.3390/ijms21124507