Genetic Markers in Lung Cancer Diagnosis: A Review


Abstract
:1. Introduction
2. Genetic Markers in Diagnosis of Early-Stage Lung Cancer
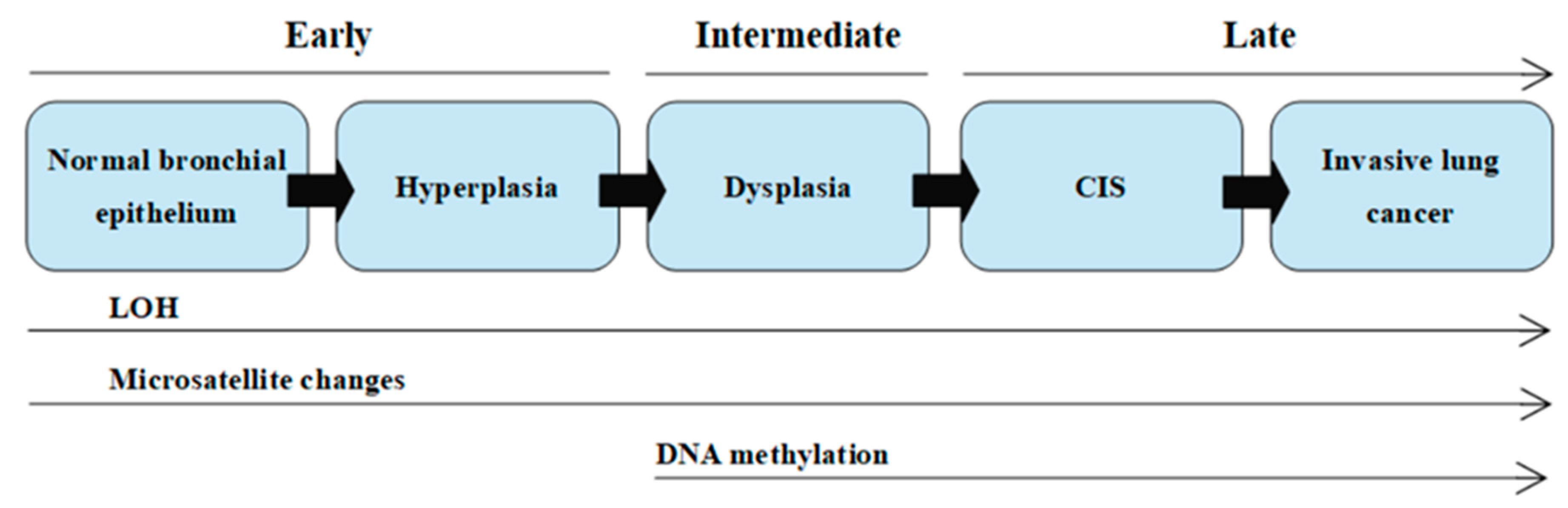
2.1. Carcinogenesis
2.2. Genetic Biomarkers
2.3. Liquid Biopsy

{kind=link}
| Type of Specimen | ADVANTAGES | DISADVANTAGES | Examples of Molecular Markers | Ref. |
|---|---|---|---|---|
| Small Histopathological Specimens *,** Cytology Specimens *,** |
|
|
| [24,30,31,32,33,34] |
| Liquid biopsy |
|
|
| [24,35,36] |
3. Advancement of Molecular Strategies and Techniques Used to Identify Lung Cancer Genetic Markers
3.1. Genomics
- (a)
- Enzymatic DNA restriction—there are four classes of restriction enzymes. The most commonly exploited are the enzymes belonging to class II, which require only ions of Mg2+ to recognize their target DNA sequence and cleave it. The usage of different combinations of restriction enzymes allows us to characterize and manipulate DNA in fundamental DNA technology approaches such as cloning or mapping [42,43].
- (b)
- Nucleic acid hybridization—in situ hybridization (ISH) uses labeled nucleic acid probes to detect specific DNA or RNA targets in tissue sections, intact cells, or chromosomes. The basic principle underlying ISH is the ability of single-stranded DNA or RNA to anneal specifically to a complementary sequence and form a double-stranded hybrid. Nucleic acid hybridization is the foundation of Southern or Northern blot hybridization and microarray technology [44,45,46]. The development of microarray technology (also known as DNA microarrays, DNA chips) is connected with the transition of molecular biology into postgenomic era by enabling large-scale genotyping and gene expression profiling [47,48].
- (c)
- Polymerase chain reaction (PCR)—PCR allows for the exponential amplification of specific targeted genetic loci in a reaction mixture containing DNA primers, deoxynucleotides (dNTPs), and DNA polymerases. PCR is a qualitative technique to amplify and copy a targeted area of extracted DNA a million to a billion-fold over [49]. In the course of time, modifications and advances in this molecular diagnostics technique enabled the relative or even absolute quantification of DNA through the usage of quantitative real-time PCR (qPCR) or partition-based PCR techniques, such as droplet digital PCR (ddPCR) [27,49]. The exploitation of PCR in quantitative DNA analysis is leading to the increased clinical usefulness of PCR for a broad range of applications; PCR is used in a variety of methods, such as allele-specific PCR-based methods or mutation screening methods, including melting curve analysis, that are used in the analysis of mutations sequences. PCR is also used in pyrosequencing and next-generation sequencing (NGS) as a pre-step that provides the sequencing of the generated PCR products [17],
- (d)
- Fluorescence-based methods—the use of hybridizing fluorescent-labeled probes is one of the advancements in cytogenetics. Fluorescence-based methods include the fluorescence in situ hybridization (FISH) and comparative genomic hybridization (CGH) techniques, which detect large-scale amplifications and deletions. FISH uses specific fluorescent probes that bind to nucleic acid sequences with a high degree of sequence complementarity, helping one to localize these DNA sequences on chromosomes. Microarray-based CGH (array CGH) enables us genome-wide screening for chromosomal imbalances on the basis of genomic DNA hybridization to complement probes that are immobilized on a slide [17,23,49].
3.2. Understanding of Molecular Pathology of Lung Cancer
- WGS and WES to find novel mutations in so far unreported gene loci;
- paired-end, mate-pair sequencing to identify structural variations;
- targeted sequencing for mutation discovery and validation;
- transcriptome sequencing for the quantification of gene expression and discovery of transcribed mutations;
- small RNA-sequencing to microRNA profiling;
3.3. Epigenomics
4. Genomic and Epigenomic Changes in Lung Cancer Diagnosis
4.1. Oncogenes and Tumor Suppressor Genes in Lung Cancer
4.2. Microsatellite Markers
4.3. Epigenetic Changes in Lung Cancer
4.4. MicroRNAs in Lung Cancer Diagnosis
5. Lung Cancer Genetic Heterogeneity
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5caC | 5-carboxylcytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| AC/ADC | Adenocarcinoma |
| ALK | Anaplastic lymphoma kinase |
| AUC | Area under the receiver operating characteristic (ROC) curve |
| BAL | Bronchoalveolar lavage |
| BRAF | B-raf proto-oncogene |
| BS-Seq | Bisulfite conversion followed by sequencing |
| CEA | Carcinoembryonic antigen |
| cfDNA | Cell-free circulating DNA |
| CGH | Comparative genomic hybridization |
| ChIP | Chromatin immunoprecipitation |
| CIS | Carcinoma in situ |
| CNV | Copy number variation |
| CTC | Circulating tumor cell |
| ctDNA | Cell-free tumor DNA |
| ddPCR | Droplet digital polymerase chain reaction |
| DFS | Disease free survival |
| DNMT | DNA methyltransferase enzyme |
| dNTP | Deoxynucleotide |
| DR | Death receptor |
| EGFR | Epidermal growth factor receptor |
| ELISA | Enzyme-linked immunosorbent assay |
| ERBB2 | Erb-B2 receptor tyrosine kinase 2 |
| FHIT | Fragile histidine triad diadenosine triphosphatase |
| FISH | Fluorescence in situ hybridization |
| HER2 | Human epidermal growth factor receptor 2 |
| HNPCC | Hereditary non-polyposis colon cancer |
| HPLC | High-performance liquid chromatography |
| HUGO | Human Genome Organization |
| IHC | Immunohistochemistry |
| ISH | In situ hybridization |
| KRAS | Kirsten rat sarcoma viral oncogene |
| LDCT | Low-dose computer tomography |
| LOH | Loss of heterogeneity |
| Mb | Megabase |
| MeFISH | Methylation-specific fluorescence in situ hybridization |
| MET | Mesenchymal-epithelial transition factor |
| MLPA | Multiplex ligation-dependent probe amplification |
| MSI | Microsatellite instability |
| NA | Not available |
| NGS | Next-generation sequencing |
| NSCLC | Non-small cell lung carcinoma/Non-small cell lung cancer |
| NSE | Neuron-specific enolase |
| OS | Overall survival |
| PCR | Polymerase chain reaction |
| PI | Proximal-inflammatory |
| PP | Proximal-proliferative |
| PPV | Positive predictive value |
| PTM | Posttranslational modification |
| qPCR | Quantitative real-time polymerase chain reaction |
| RET | Rearranged during transfection |
| RISC | RNA-induced silencing complex |
| ROC | Receiver operating characteristic |
| ROS1 | C-ros oncogene 1 |
| RT-PCR | Reverse-transcription polymerase chain reaction |
| SCC | Squamous cell carcinoma/Squamous cell cancer |
| SCCA | Squamous cell carcinoma antigen |
| SCLC | Small cell lung carcinoma/Small cell lung cancer |
| SCNA | Somatic copy number alteration |
| SNP | Single nucleotide polymorphism |
| SNV | Single nucleotide variant |
| TCGA | The Cancer Genome Atlas |
| TKI | Tyrosine kinase inhibitor |
| TMB | Tumor mutation burden |
| TNM | TNM Classification of Malignant Tumors (tumor-lymph nodes-metastasis) |
| TRU | Terminal respiratory unit |
| WES | Whole-exome sequencing |
| WGS | Whole-genome sequencing |
References
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahal, Z.; El Nemr, S.; Sinjab, A.; Chami, H.; Tfayli, A.; Kadara, H. Smoking and Lung Cancer: A Geo-Regional Perspective. Front. Oncol. 2017, 7, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.L.; Paz-Ares, L. Lung Cancer: Current Therapies and New Targeted Treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Xi, K.X.; Zhang, X.W.; Yu, X.Y.; Wang, W.D.; Xi, K.X.; Chen, Y.Q.; Wen, Y.S.; Zhang, L.J. The role of plasma miRNAs in the diagnosis of pulmonary nodules. J. Thorac. Dis. 2018, 10, 4032–4041. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Kong, H.; Hou, Y.; Ge, D.; Huang, W.; Ou, J.; Yang, D.; Zhang, L.; Wu, G.; Song, Y.; et al. Two Plasma microRNA Panels For Diagnosis and Subtype Discrimination of Lung Cancer. Lung Cancer. 2018, 123, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Jakubek, Y.; Lang, W.; Vattathil, S.; Garcia, M.; Xu, L.; Huang, L.; Yoo, S.Y.; Shen, L.; Lu, W.; Chow, C.W.; et al. Genomic Landscape Established by Allelic Imbalance in the Cancerization Field of a Normal Appearing Airway. Cancer Res. 2016, 76, 3676–3683. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; Franklin, W.A.; Gazdar, A.F.; Bunn, P.A. Early Detection of Lung Cancer: Clinical Perspectives of Recent Advances in Biology and Radiology. Clin. Cancer Res. 2001, 7, 5–22. [Google Scholar]
- Santarpia, M.; Liguori, A.; D’Aveni, A.; Karachaliou, N.; Gonzalez-Cao, M.; Daffinà, M.G.; Lazzari, C.; Altavilla, G.; Rosell, R. Liquid biopsy for lung cancer early detection. J. Thorac. Dis. 2018, 10, S882–S897. [Google Scholar] [CrossRef] [Green Version]
- Gazdar, A.F.; Brambilla, E. Preneoplasia of lung cancer. Cancer Biomark. 2010, 9, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Jassem, E.; Szymanowska, A.; Siemińska, A.; Jassem, J. Palenie tytoniu a rak płuca. Pneumonol. Alergol. Pol. 2009, 77, 469–473. [Google Scholar]
- Wistuba, I.I.; Behrens, C.; Milchgrub, S.; Bryant, D.; Hung, J.; Minna, J.D.; Gazdar, A.F. Sequential Molecular Abnormalities Are Involved in the Multistage Development of Squamous Cell Lung Carcinoma. Oncogene 1999, 18, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potempa, M.; Jonczyk, P.; Zalewska-Ziob, M. Molekularne uwarunkowania raka płuca. Onkol. Prak. Klin. 2014, 10, 199–211. [Google Scholar]
- Müllauer, L. Next generation sequencing: Clinical applications in solid tumours. Memo 2017, 10, 244–247. [Google Scholar] [CrossRef] [Green Version]
- Hubers, A.J.; Prinsen, C.F.; Sozzi, G.; Witte, B.I.; Thunnissen, E. Molecular sputum analysis for the diagnosis of lung cancer. Br. J. Cancer 2013, 109, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Ahrendt, S.A.; Chow, J.T.; Xu, L.; Yang, S.C.; Eisenberger, C.F.; Esteller, M.; Herman, J.G.; Wu, L.; Decker, A.; Jen, J.; et al. Molecular Detection of Tumor Cells in Bronchoalveolar Lavage Fluid From Patients With Early Stage Lung Cancer. J. Natl. Cancer Inst. 1999, 91, 332–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadara, H.; Scheet, P.; Wistuba, I.I. Early Events in the Molecular Pathogenesis of Lung Cancer. Cancer Prev. Res. (Phila.) 2016, 9, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Starostik, P. Clinical mutation assay of tumors: New developments. Anti-Cancer Drugs. 2017, 28, 1–10. [Google Scholar] [CrossRef]
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 160–169. [Google Scholar] [CrossRef]
- Ou, S.I.; Nagasaka, M.; Zhu, V.W. Liquid Biopsy to Identify Actionable Genomic Alterations. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 978–997. [Google Scholar] [CrossRef]
- Zhang, X.; Liang, Z.; Wang, S.; Lu, S.; Song, Y.; Cheng, Y.; Ying, J.; Liu, W.; Hou, Y.; Li, Y.; et al. Application of next-generation sequencing technology to precision medicine in cancer: Joint consensus of the Tumor Biomarker Committee of the Chinese Society of Clinical Oncology. Cancer Biol. Med. 2019, 16, 189–204. [Google Scholar]
- García-Giménez, J.L.; Seco-Cervera, M.; Tollefsbol, T.O.; Romá-Mateo, C.; Peiró-Chova, L.; Lapunzina, P.; Pallardó, F.V. Epigenetic biomarkers: Current strategies and future challenges for their use in the clinical laboratory. Crit. Rev. Clin. Lab. Sci. 2017, 54, 529–550. [Google Scholar] [CrossRef] [PubMed]
- Lis, P.; Niczyj-Raucy, M.; Lis, M. The molecular basis of cancer and genetic methods of its diagnosis. Nat. J. (Opole) 2011, 44, 92–119. [Google Scholar]
- Pass, H.I.; Beer, D.G.; Joseph, S.; Massion, P. Biomarkers and Molecular Testing for Early Detection, Diagnosis, and Therapeutic Prediction of Lung Cancer. Thorac. Surg. Clin. 2013, 23, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Lunardi, F.; Pezzuto, F.; Fortarezza, F.; Vuljan, S.E.; Marquette, C.; Hofman, P. Are There New Biomarkers in Tissue and Liquid Biopsies for the Early Detection of Non-Small Cell Lung Cancer? J. Clin. Med. 2019, 8, 414. [Google Scholar] [CrossRef] [Green Version]
- Hassanein, M.; Callison, J.C.; Callaway-Lane, C.; Aldrich, M.C.; Grogan, E.L.; Massion, P.P. The state of molecular biomarkers for the early detection of lung cancer. Cancer Prev. Res. (Phila.) 2012, 5, 992–1006. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L. Molecular diagnostics of lung cancer in the clinic. Transl. Lung Cancer Res. 2017, 6, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Liquid Biopsy and Droplet Digital PCR Offer Improvements for Lung Cancer Testing. Available online: http://archive.vn/2020.06.26-095203/https://www.mlo-online.com/continuing-education/article/13017057/liquid-biopsy-and-droplet-digital-pcr-offer-improvements-for-lung-cancer-testing (accessed on 25 April 2020).
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Sholl, L.M.; Aisner, D.L.; Allen, T.C.; Beasley, M.B.; Cagle, P.T.; Capelozzi, V.L.; Dacis, S.; Hariri, L.P.; Kerr, K.M.; Lantuejoul, S.; et al. Liquid Biopsy in Lung Cancer: A Perspective From Members of the Pulmonary Pathology Society. Arch. Pathol. Lab. Med. 2016, 140, 825–829. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yu, B.; Ng, C.; Mercorella, B.; Selinger, C.; O′Toole, S.; Cooper, W. The suitability of small biopsy and cytology specimens for EGFR and other mutation testing in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 119–125. [Google Scholar]
- Bubendorf, L.; Lantuejoul, S.; de Langen, A.; Thunnissen, E. Non-small cell lung carcinoma: Diagnostic difficulties in small biopsies and cytological specimens. Eur. Respir. Rev. 2017, 26, 170007. [Google Scholar] [CrossRef]
- Sigel, C.; Moreira, A.; Travis, W.; Zakowski, M.; Thornton, R.; Riely, G.; Rekhtman, N. Subtyping of Non-small Cell Lung Carcinoma: A Comparison of Small Biopsy and Cytology Specimens. J. Thorac. Oncol. 2011, 6, 1849–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, W.; Brambilla, E.; Noguchi, M.; Nicholson, A.; Geisinger, K.; Yatabe, Y.; Ishikawa, Y.; Wistuba, I.; Flieder, D.; Franklin, W.; et al. Diagnosis of Lung Cancer in Small Biopsies and Cytology: Implications of the 2011 International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society Classification. Arch. Pathol. Lab. Med. 2013, 137, 668–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, A.; Barnes, D.; Troy, L. Diagnosing Lung Cancer: The Complexities of Obtaining a Tissue Diagnosis in the Era of Minimally Invasive and Personalised Medicine. J. Clin. Med. 2018, 7, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sromek, M.; Glogowski, M.; Chechlinska, M.; Kulinczak, M.; Szafron, L.; Zakrzewska, K.; Owczarek, J.; Wiśniewski, P.; Wlodarczyk, R.; Talarek, L.; et al. Changes in plasma miR-9, miR-16, miR-205 and miR-486 levels after non-small cell lung cancer resection. Cell Oncol. (Dordr.) 2017, 40, 529–536. [Google Scholar] [CrossRef]
- Johann, D.; Steliga, M.; Shin, I.; Yoon, D.; Arnaoutakis, K.; Hutchins, L.; Liu, M.; Liem, J.; Walker, K.; Pereira, A.; et al. Liquid biopsy and its role in an advanced clinical trial for lung cancer. Exp. Biol. Med. 2018, 243, 262–271. [Google Scholar] [CrossRef]
- Mellert, H.; Jackson, L.; Pestano, G. Performance verification of a plasma-based PD-L1 test that reliably measures mRNA expression from patients with NCSLC. J. Clin. Oncol. 2018, 36, 156. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Daniels, M.G.; Bowman, R.V.; Yang, I.A.; Govindan, R.; Fong, K.M. An emerging place for lung cancer genomics in 2013. J. Thorac. Dis. 2013, 5, S491–S497. [Google Scholar]
- McDermott, U.; Downing, J.R.; Stratton, M.R. Genomics and the continuum of cancer care. N. Engl. J. Med. 2011, 364, 340–350. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Shah, P.; Pandey, D.; Kumar, A. Molecular Diagnostic Technology. In Biotechnology in Medicine and Agriculture Principles and Practices; Kumar, A., Pareek, A., Gupta, S.M., Eds.; I K International Publishing House: New Delhi, India, 2012; pp. 368–402. [Google Scholar]
- Bal, J.; Wiszniewska, J.; Wiszniewski, W. Metody analizy genomu. In Genetyka Medyczna i Molekularna; Bal, J., Ed.; PWN: Warszawa, Poland, 2017; pp. 101–118. [Google Scholar]
- DI Felice, F.; Micheli, G.; Camilloni, G. Restriction enzymes and their use in molecular biology: An overview. J. Biosci. 2019, 44, 38. [Google Scholar] [CrossRef]
- Cowlen, M.S. Nucleic Acid Hybridization and Amplification In Situ. In Molecular Diagnostics. Pathology and Laboratory Medicine; Coleman, W.B., Tsongalis, G.J., Eds.; Humana Press: Totowa, NJ, USA, 1997; pp. 163–191. [Google Scholar]
- Netzer, K.O. Hybridization Methods (Southern and Northern Blotting). In Techniques in Molecular Medicine; Hildebrandt, F., Igarashi, P., Springer Lab Manual, Eds.; Springer: Berlin, Germany, 1999; pp. 126–147. [Google Scholar]
- Blohm, D.H.; Guiseppi-Elie, A. New developments in microarray technology. Curr. Opin. Biotech. 2001, 12, 41–47. [Google Scholar] [CrossRef]
- Jarząb, B.; Gubała, E.; Lange, D. Mikromacierze DNA i profil ekspresji genów raka brodawkowatego tarczycy. Endokrynol. Pol. 2005, 3, 294–301. [Google Scholar]
- Mirski, T.; Bartoszcze, M.; Bielawska-Drózd, A.; Gryko, R.; Kocik, J.; Niemcewicz, M.; Chomiczewski, K. Microarrays—New Possibilities for Detecting Biological Factors Hazardous for Humans and Animals, and for Use in Environmental Protection. Ann. Agric. Environ. Med. 2016, 23, 30–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, K.; Singh, A.; Butzmann, A.; Jangam, D.; Ohgami, R.S. Molecular genetic testing methodologies in hematopoietic diseases: Current and future methods. Int. J. Lab. Hematol. 2019, 41, 102–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Kuo, F.C.; Mar, B.G.; Lindsley, R.C.; Lindeman, N.I. The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood 2017, 130, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Vendrell, J.A.; Grand, D.; Rouquette, I.; Costes, V.; Icher, S.; Selves, J.; Larrieux, M.; Barbe, A.; Brousset, P.; Solassol, J. High-throughput detection of clinically targetable alterations using next-generation sequencing. Oncotarget 2017, 8, 40345–40358. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Kim, J.W. Principles of Genetic Counseling in the Era of Next-Generation Sequencing. Ann. Lab. Med. 2018, 38, 291–295. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, L.; Roberts, R.; Tong, W. Toward Clinical Implementation of NextGeneration Sequencing-Based Genetic Testing in Rare Diseases: Where Are We? Trends Genet. 2019, 35, 852–867. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Shim, H.S. Detection of Targetable Genetic Alterations in Korean Lung Cancer Patients: A Comparison Study of Single-Gene Assays and Targeted Next-Generation Sequencing. Cancer Res. Treat 2020, 52, 543–551. [Google Scholar] [CrossRef]
- Dama, E.; Melocchi, V.; Colangelo, T.; Cuttano, R.; Bianchi, F. Deciphering the Molecular Profile of Lung Cancer: New Strategies for the Early Detection and Prognostic Stratification. J. Clin. Med. 2019, 18, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietel, M.; Jöhrens, K.; Laffert, M.; Hummel, M.; Bläker, H.; Müller, B.M.; Lehmann, A.; Denkert, C.; Heppner, F.L.; Koch, A.; et al. Predictive molecular pathology and its role in targeted cancer therapy: A review focussing on clinical relevance. Cancer Gene Ther. 2013, 20, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, M.; Roy, S.; Bittar, H.T.; Dacic, S. Next-Generation Sequencing Approach to Non-Small Cell Lung Carcinoma Yields More Actionable Alterations. Arch. Pathol. Lab. Med. 2018, 142, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sands, J.M.; Nguyen, T.; Shivdasani, P.; Sacher, A.G.; Cheng, M.L.; Alden, R.S.; Jänne, P.A.; Kuo, F.C.; Oxnard, G.R.; Sholl, L.M. Next-generation sequencing informs diagnosis and identifies unexpected therapeutic targets in lung squamous cell carcinomas. Lung Cancer 2020, 140, 35–41. [Google Scholar] [CrossRef]
- Fernandes, M.G.O.; Jacob, M.; Martins, N.; Moura, C.S.; Guimarães, S.; Reis, J.P.; Justino, A.; Pina, M.J.; Cirnes, L.; Sousa, C.; et al. Targeted Gene Next-Generation Sequencing Panel in Patients with Advanced Lung Adenocarcinoma: Paving the Way for Clinical Implementation. Cancers 2019, 11, 1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Q.; Wagner, U.; Kurt, H.; Molinari, F.; Cathomas, G.; Komminoth, P.; Barman-Aksözen, J.; Schneider-Yin, X.; Rey, J.P.; Vassella, E.; et al. Multi-laboratory proficiency testing of clinical cancer genomic profiling by next-generation sequencing. Pathol. Res. Pract. 2018, 214, 957–963. [Google Scholar] [CrossRef]
- McBride, C.M.; Koehly, L.M. Imagining roles for epigenetics in health promotion research. J. Behav. Med. 2017, 40, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, T.; Koyama, K.; Hayashi, G.; Okamoto, A. Chemistry-Driven Epigenetic Investigation of Histone and DNA Modifications. Chem. Rec. 2018, 18, 1727–1744. [Google Scholar] [CrossRef]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Tsou, J.A.; Hagen, J.A.; Carpenter, C.L.; Laird-Offringa, I.A. DNA methylation analysis: A powerful new tool for lung cancer diagnosis. Oncogene 2002, 21, 5450–5461. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Lee, H.J.; Smallwood, S.A.; Kelsey, G.; Reik, W. Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016, 18, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopczyński, P.; Krawczyński, M.R. The Role of Oncogenes and Tumor Suppressor Genes in Oncogenesis. Now. Lekarskie 2012, 81, 679–681. [Google Scholar]
- Sholl, L.M.; Aisner, D.L.; Varella-Garcia, M.; Berry, L.D.; Dias-Santagata, D.; Wistuba, I.I.; Chen, H.; Fujimoto, J.; Kugler, K.; Franklin, W.A.; et al. Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2015, 10, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Devarakonda, S.; Rotolo, F.; Tsao, M.S.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef]
- NCCN. National Comprehensive Cancer Network Guidelines. Available online: http://archive.today/2020.06.26-094846/https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 10 May 2020).
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch. Pathol. Lab. Med. 2018, 142, 321–346. [Google Scholar]
- Kim, E.Y.; Lee, S.H.; Kim, A.; Kim, T.; Chang, Y.S. Tumor clonal status predicts clinical outcomes of lung adenocarcinoma with EGFR-TKI sensitizing mutation. J. Cancer 2019, 10, 5549–5556. [Google Scholar] [CrossRef]
- Garrido, P.; Conde, E.; de Castro, J.; Gómez-Román, J.; Felip, E.; Pijuan, L.; Isla, D.; Sanz, J.; Paz-Ares, L.; López-Ríos, F. Updated guidelines for predictive biomarker testing in advanced non-small-cell lung cancer: A National Consensus of the Spanish Society of Pathology and the Spanish Society of Medical Oncology. Transl. Oncol. 2020, 22, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Popper, H.; Tímár, J.; Ryska, A.; Olszewski, W. Minimal requirements for the molecular testing of lung cancer. Transl. Lung Cancer Res. 2014, 3, 301–304. [Google Scholar] [PubMed] [Green Version]
- Planchard, D. Other Oncogenic Drivers (BRAF, MET, RET, HER2, NTRK). Available online: http://splf.fr/wp-content/uploads/2018/10/S14-3.pdf (accessed on 22 May 2020).
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebb, D.G.; Agulnik, J.; Albadine, R.; Banerji, S.; Bigras, G.; Butts, C.; Couture, C.; Cutz, J.C.; Desmeules, P.; Ionescu, D.N.; et al. Crizotinib inhibition of ROS1-positive tumours in advanced non-small-cell lung cancer: A Canadian perspective. Curr. Oncol. 2019, 26, e551–e557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.; Jung, H.A.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Ahn, M.J. Clinical Characteristics and Outcomes of Non-small Cell Lung Cancer Patients with HER2 Alterations in Korea. Cancer Res. Treat. 2020, 52, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Janke, F.; Bozorgmehr, F.; Wrenger, S.; Dietz, S.; Heussel, C.; Heussel, G.; Silva, C.; Rheinheimer, S.; Feisst, M.; Thomas, M.; et al. Novel Liquid Biomarker Panels for A Very Early Response Capturing of NSCLC Therapies in Advanced Stages. Cancers 2020, 12, 954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Wang, X.; Tian, L.; Che, G. Microsatellite alteration in multiple primary lung cancer. J. Thorac. Dis. 2014, 6, 1499–1505. [Google Scholar] [PubMed]
- The Cancer Genome Atlas Network; (Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al.) Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Sozzi, G.; Musso, K.; Ratcliffe, C. Detection of Microsatellite Alternations in Plasma DNA of Non-Small Cell Lung Cancer Patients: A Prospect for Early Diagosis. Clin. Cancer Res. 1999, 5, 2689–2692. [Google Scholar]
- Pęcherzewska, R.; Nawrot, B. FHIT—Tumor Suppressor Protein Involved in Induction of Apoptosis and Cell Cycle Regulation. Postepy Biochem. 2009, 55, 66–75. [Google Scholar]
- Łaczmańska, I.; Ślęzak, R. Etiologia i znaczenie kliniczne miejsc kruchych w chromosomach człowieka. Laboratory Diagnostics 2010, 46, 81–86. [Google Scholar]
- Duruisseaux, M.; Esteller, M. Lung cancer epigenetics: From knowledge to applications. Semin. Cancer Biol. 2018, 51, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, W.A.; Divine, K.K.; Saccomanno, G.; Gilliland, F.D.; Baylin, S.B.; Herman, J.G.; Belinsky, S.A. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000, 60, 5954–5958. [Google Scholar] [PubMed]
- Ooki, A.; Maleki, Z.; Tsay, J.J.; Goparaju, C.; Brait, M.; Turaga, N.; Nam, H.S.; Rom, W.N.; Pass, H.I.; Sidransky, D.; et al. A Panel of Novel Detection and Prognostic Methylated DNA Markers in Primary Non-Small Cell Lung Cancer and Serum DNA. Clin. Cancer Res. 2017, 23, 7141–7152. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Zhang, H.; Lu, S.; Wu, Z.; Zhou, L.; Cheng, Z.; Bai, Y.; Zhao, J.; Zhang, Q.; Mao, H. Quantitative assessment of gene promoter methylation in non-small cell lung cancer using methylation-sensitive high-resolution melting. Oncol. Lett. 2018, 15, 7639–7648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Li, J.; Zhang, C.; Hong, Q.; Jiang, D.; Ye, M.; Duan, S. Distinguishing Lung Adenocarcinoma from Lung Squamous Cell Carcinoma by Two Hypomethylated and Three Hypermethylated Genes: A Meta-Analysis. PLoS ONE 2016, 11, e0149088. [Google Scholar] [CrossRef] [Green Version]
- Filip, A. MikroRNA: Nowe mechanizmy regulacji ekspresji genów. Postępy Biochemii. 2007, 53, 413–419. [Google Scholar]
- Aiso, T.; Ohtsuka, K.; Ueda, M.; Karita, S.; Yokoyama, T.; Takata, S.; Matsuki, N.; Kondo, H.; Takizawa, H.; Okada, A.A.; et al. Serum levels of candidate microRNA diagnostic markers differ among the stages of non-small-cell lung cancer. Oncol. Lett. 2018, 16, 6643–6651. [Google Scholar] [CrossRef]
- Ulivi, P.; Petracci, E.; Marisi, G.; Baglivo, S.; Chiari, R.; Billi, M.; Canale, M.; Pasini, L.; Racanicchi, S.; Vagheggini, A.; et al. Prognostic Role of Circulating miRNAs in Early-Stage Non-Small Cell Lung Cancer. J. Clin Med. 2019, 23, 131. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, F.; Nicassio, F.; Marzi, M.; Belloni, E.; Dall′olio, V.; Bernard, L.; Pelosi, G.; Maisonneuve, P.; Veronesi, G.; Di Fiore, P.P. A serum circulating miRNA diagnostic test to identify asymptomatic high-risk individuals with early stage lung cancer. EMBO. Mol. Med. 2011, 3, 495–503. [Google Scholar] [CrossRef]
- Xing, L.; Todd, N.W.; Yu, L.; Frang, H.; Jiang, F. Early Detection of Squamous Cell Lung Cancer in Sputum by a Panel of microRNA Markers. Mod. Pathol. 2010, 23, 1157–1164. [Google Scholar] [CrossRef]
- Yu, L.; Todd, N.W.; Xing, L.; Xie, Y.; Zhang, H.; Liu, Z.; Fang, H.; Zhang, J.; Katz, R.L.; Jiang, F. Early detection of lung adenocarcinoma in sputum by a panel of microRNA markers. Int. J. Cancer 2010, 127, 2870–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Haddadin, S.; Wang, Y.; Gu, L.Q.; Perry, M.C.; Freter, C.E.; Wang, M.X. Plasma microRNAs as novel biomarkers for early detection of lung cancer. Int. J. Clin. Exp. Pathol. 2011, 4, 575–586. [Google Scholar] [PubMed]
- Hennessey, P.T.; Sanford, T.; Choudhary, A.; Mydlarz, W.W.; Brown, D.; Adai, A.T.; Ochs, M.F.; Ahrendt, S.A.; Mambo, E.; Califano, J.A. Serum microRNA biomarkers for detection of non-small cell lung cancer. PLoS ONE 2012, 7, e32307. [Google Scholar] [CrossRef]
- Heegaard, N.H.; Schetter, A.J.; Welsh, J.A.; Yoneda, M.; Bowman, E.D.; Harris, C.C. Circulating micro-RNA expression profiles in early stage nonsmall cell lung cancer. Int. J. Cancer 2012, 130, 1378–1386. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhu, S.; Tao, Z.; Ye, S. High circulating miR-18a, miR-20a, and miR-92a expression correlates with poor prognosis in patients with non-small cell lung cancer. Cancer Med. 2018, 7, 21–31. [Google Scholar] [CrossRef]
- Yan, H.Z.; Wang, W.; Du, X.; Jiang, X.D.; Lin, C.Y.; Guo, J.L.; Zhang, J. The Expression and Clinical Significance of miRNA-99a and miRNA-224 in Non-Small-Cell Lung Cancer. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1545–1552. [Google Scholar]
- Szczyrek, M.; Kuźnar-Kamińska, B.; Grenda, A.; Krawczyk, P.; Sawicki, M.; Głogowski, M.; Balicka, G.; Rolska-Kopińska, A.; Nicoś, M.; Jakimiec, M.; et al. Diagnostic Value of Plasma Expression of microRNAs Complementary to Drosha and Dicer in Lung Cancer Patients. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3857–3866. [Google Scholar]
- Beganoyic, S. Clinical Significance of the Kras Mutation. Bosn. J. Basic Med. Sci. 2009, 9, S17–S20. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. CHS. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Ramón y Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar]
- Lin, J.; Shaw, A. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016, 2, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricordel, C.; Friboulet, L.; Facchinetti, F.; Soria, J. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann. Oncol. 2018, 29, i28–i37. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Cui, J.; Schrock, A.; Goldberg, M.; Zhu, V.; Albacker, L.; Stephens, P.; Miller, V.; Ali, S. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/G and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Zhou, F.; Shen, W.; Jiang, T.; Wu, X.; Tong, X.; Shao, Y.; Qin, S.; Zhou, C. Novel Mutations on EGFR Leu792 Potentially Correlate to Acquired Resistance to Osimertinib in Advanced NSCLC. J. Thorac. Oncol. 2017, 12, e65–e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kim, T.; Kim, D.; Go, H.; Keam, B.; Lee, S.; Ku, J.; Chung, D.; Heo, D. Heterogeneity of Genetic Changes Associated with Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancer. J. Thorac. Oncol. 2013, 8, 415–422. [Google Scholar] [CrossRef] [Green Version]

| Mutation Detection Techniques | Variant Types | |
|---|---|---|
| SNVs | CNVs | |
| Single-gene assays: | ||
| Sanger sequencing | + | - |
| pyrosequencing | + | - |
| allele-specific PCR | + | - |
| single base extension | + | - |
| multiplex ligation-dependent probe amplification | + | copy number only |
| mass spectrometry | + | - |
| Gene-panel assays: | ||
| amplicon-based panels | - | + |
| hybrid capture sequencing | - | + |
| next-generation sequencing | + | + |
| Fluorescence-based methods: | ||
| fluorescence in situ hybridization | - | + |
| microarray-based CGH | - | + |
| Gene | Type of Genomic Aberrations | Frequency [%] | Currently Available Targeted Therapy * | Diagnostic Approaches | Ref. |
|---|---|---|---|---|---|
| Adenocarcinomas (ADC) | |||||
| EGFR | EGFR-TKI sensitizing mutations: EGFR exon 21, EGFR exon 19, G719X, L861Q point mutations Copy number variations (gains) | 30–40 | pemetrexed or bevacizumab therapy, afatinib, erlotinib, gefitinib, dacomitinib, osimertinib | PCR: sanger, real-time PCR, ddPCR, and NGS; IHC | [67,72,73,74,75,76,77] |
| KRAS | G12C mutation in KRAS gene | 20–30 | AMG-510 | PCR, DNA sequencing | [67,72,73,74,77] |
| MET | MET exon 14 mutation (MET ex14), skipping mutations, overexpression, amplifications | 2–5 3–4 | skipping mutations—crizotinib, tepotinib; amplifications—crizotinib, capmatinib | mutations: sanger sequencing, NGS; amplifications: FISH, PCR, real-time PCR, NGS | [67,72,73,74,76,78] |
| ALK | ALK fusions | 3–7 | crizotinib, alectinib, ceritinib, brigatinib, lorlatinib | FISH (the gold standard); ALK-IHC has become a widely used technique with two validated antibodies in lung cancer (D5F3, 5A4) | [67,72,73,74,76,77,79] |
| BRAF | V600E mutation in BRAF gene; can co-exist with KRAS mutation | 0.5–5 | trametinib, dabrafenib | PCR: sanger, real-time PCR, and NGS | [67,72,73,74,76,78] |
| ROS1 | ROS fusions | 2–3 | crizotinib | ROS1-IHC (screening) is still evolving (the use of the D4D6 rabbit monoclonal antibody) **; FISH; NGS | [67,72,73,74,76,77,80] |
| RET | RET rearrangements, gene fusion of KIF5B-RET; point mutations | 1–2 | vandetanib, cabozantinib, alectinib, BLU-667, LOXO-292 | RT-PCR is typically combined with FISH; FISH; NGS | [67,72,73,74,76,78] |
| NTRK | NTRK rearrangements, gene fusions of NTRK1 (NTRKA), NTRK2 (NTRKB), NTRK3 (NTRKC) | 1–2 | entrectinib, larotrectinib, LOXO-195, repotrectinib | NGS with a panel that includes testing for NTRK1, NTRK2, NTRK3; IHC with subsequent confirmation by FISH or NGS | [67,72,73,74,76,78] |
| HER2 *** | mutations in the kinase domain (exon 20), the most frequent is p.A775_G776insYVMA insertion amplifications, overexpressions | 1–5 2–5 | afatinib, dacomitinib, neratinib, trastuzumab, trastuzumab-emtansine, DS-8201a, poziotinib | mutations: PCR: sanger, real-time PCR and NGS; amplifications: FISH, NGS, real-time PCR | [72,74,76,78,81] |
| PTEN PDGFRA PIK3CA TP53 ERBB2 TERT CDKN2A | mutations copy number variations—gains losses | 1.7 6–7 5 52 2–5 75 7 | NA NA NA NA NA NA NA | - **** | [59,67,68] |
| Squamous cell carcinoma (SCC) | |||||
| FGFR TP53 NF1 DDR2 PDGFRA PIK3CA PTEN SOX2 CDKN2A | gene fusion of FGFR3-TACC3, mutations of FGFR1, FGFR2 tumor suppressor mutations, copy number variations (gains) mutations of NF1 point mutations of DDR2 amplification amplification tumor suppressor mutations, copy number variations (losses) amplification and copy number variation (gain) copy number variation (loss) | 23 79 10 2–3 4 15 10 8 65 15 | NA NA NA NA NA NA NA NA NA | [59,67,68] | |
| Name of the Unit | Abbreviation | Formerly | Mutations |
|---|---|---|---|
| Terminal respiratory unit | TRU | bronchioid | mutations in the EGFR gene and tumors expressing the kinase fusion; |
| Proximal-inflammatory | PI | squamoid | mutations in NF1 and TP53 genes |
| Proximal-proliferative | PP | magnoid | mutations of KRAS oncogene and inactivation of the STK11 tumor suppressor gene |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wadowska, K.; Bil-Lula, I.; Trembecki, Ł.; Śliwińska-Mossoń, M. Genetic Markers in Lung Cancer Diagnosis: A Review. Int. J. Mol. Sci. 2020, 21, 4569. https://doi.org/10.3390/ijms21134569
Wadowska K, Bil-Lula I, Trembecki Ł, Śliwińska-Mossoń M. Genetic Markers in Lung Cancer Diagnosis: A Review. International Journal of Molecular Sciences. 2020; 21(13):4569. https://doi.org/10.3390/ijms21134569
Chicago/Turabian StyleWadowska, Katarzyna, Iwona Bil-Lula, Łukasz Trembecki, and Mariola Śliwińska-Mossoń. 2020. "Genetic Markers in Lung Cancer Diagnosis: A Review" International Journal of Molecular Sciences 21, no. 13: 4569. https://doi.org/10.3390/ijms21134569
APA StyleWadowska, K., Bil-Lula, I., Trembecki, Ł., & Śliwińska-Mossoń, M. (2020). Genetic Markers in Lung Cancer Diagnosis: A Review. International Journal of Molecular Sciences, 21(13), 4569. https://doi.org/10.3390/ijms21134569