Emerging Strategies Targeting Catabolic Muscle Stress Relief


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathways Modulating Muscle Atrophy
2.1. Proteasome-Dependent Degradation
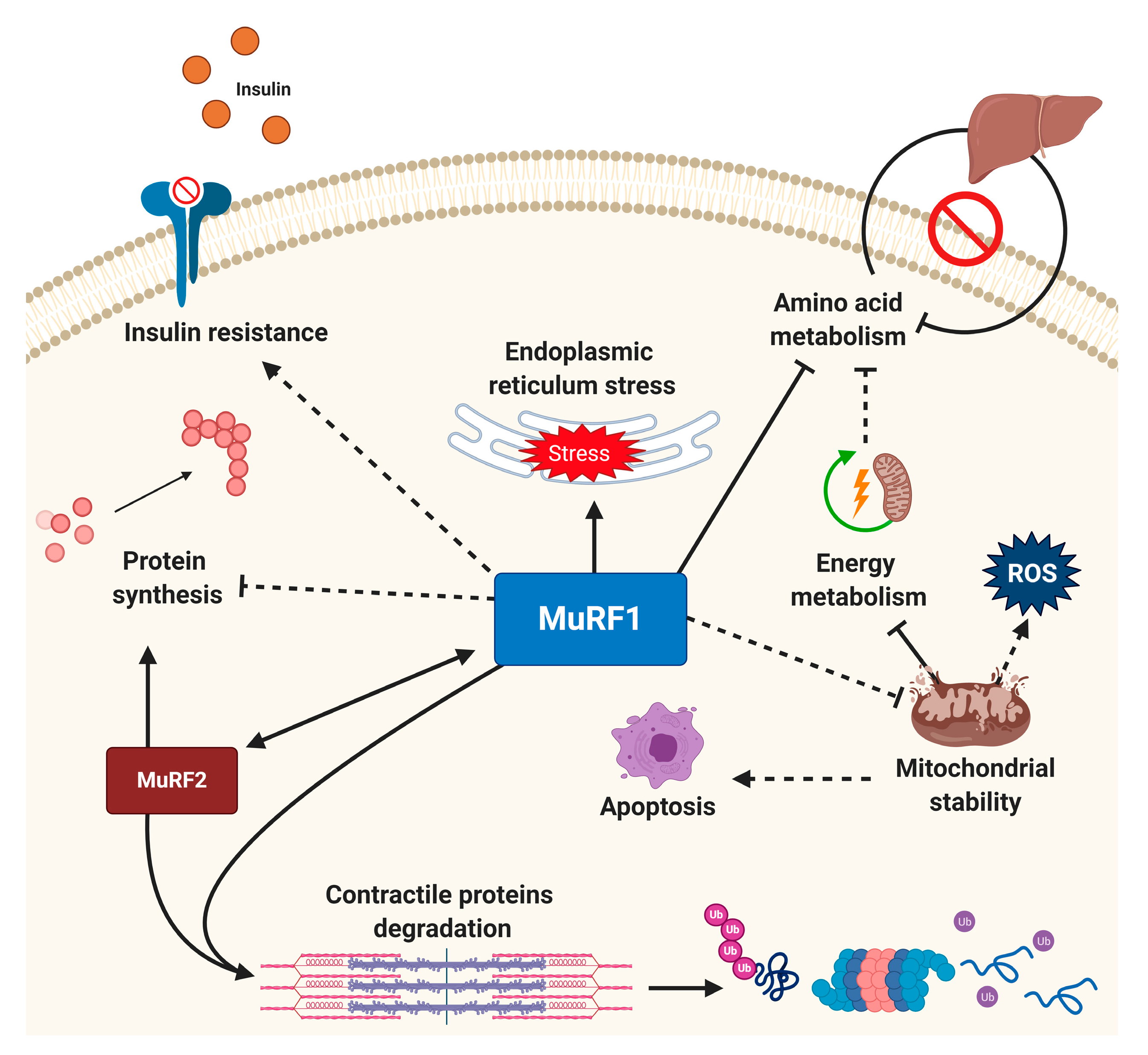
2.2. Muscle-Specific E3 Ligases: A Rate-Limiting Step in the UPS
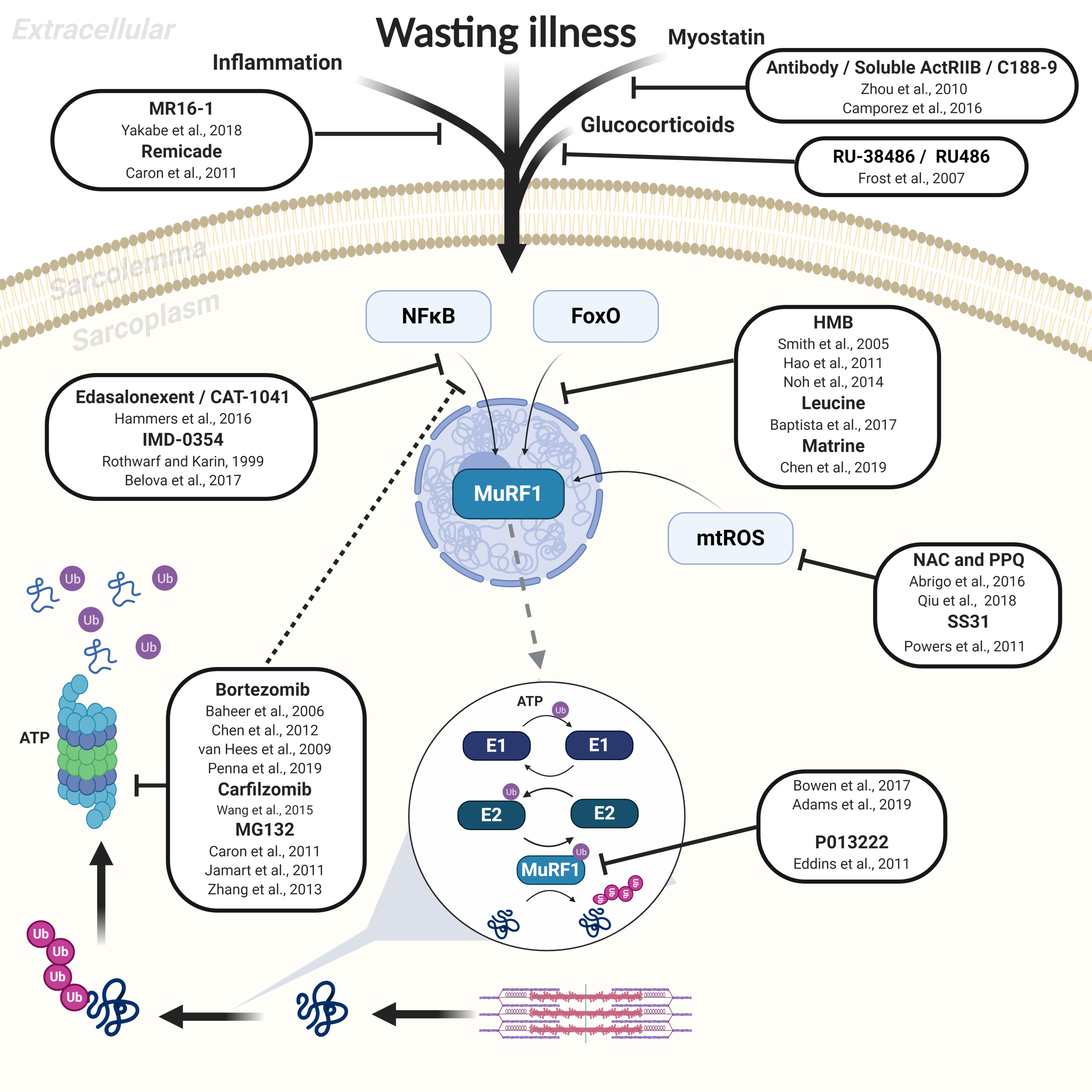
3. Therapeutic Treatments to Inhibit Muscle Atrophy
3.1. Upstream Inhibition of UPS: Inflammatory Cytokines, Growth Factors, and Transcription Factors
3.2. Downstream Inhibition of UPS via the 26S Proteasome
4. Targeted Small-Molecule Inhibition of the E3 Ligase MuRF1
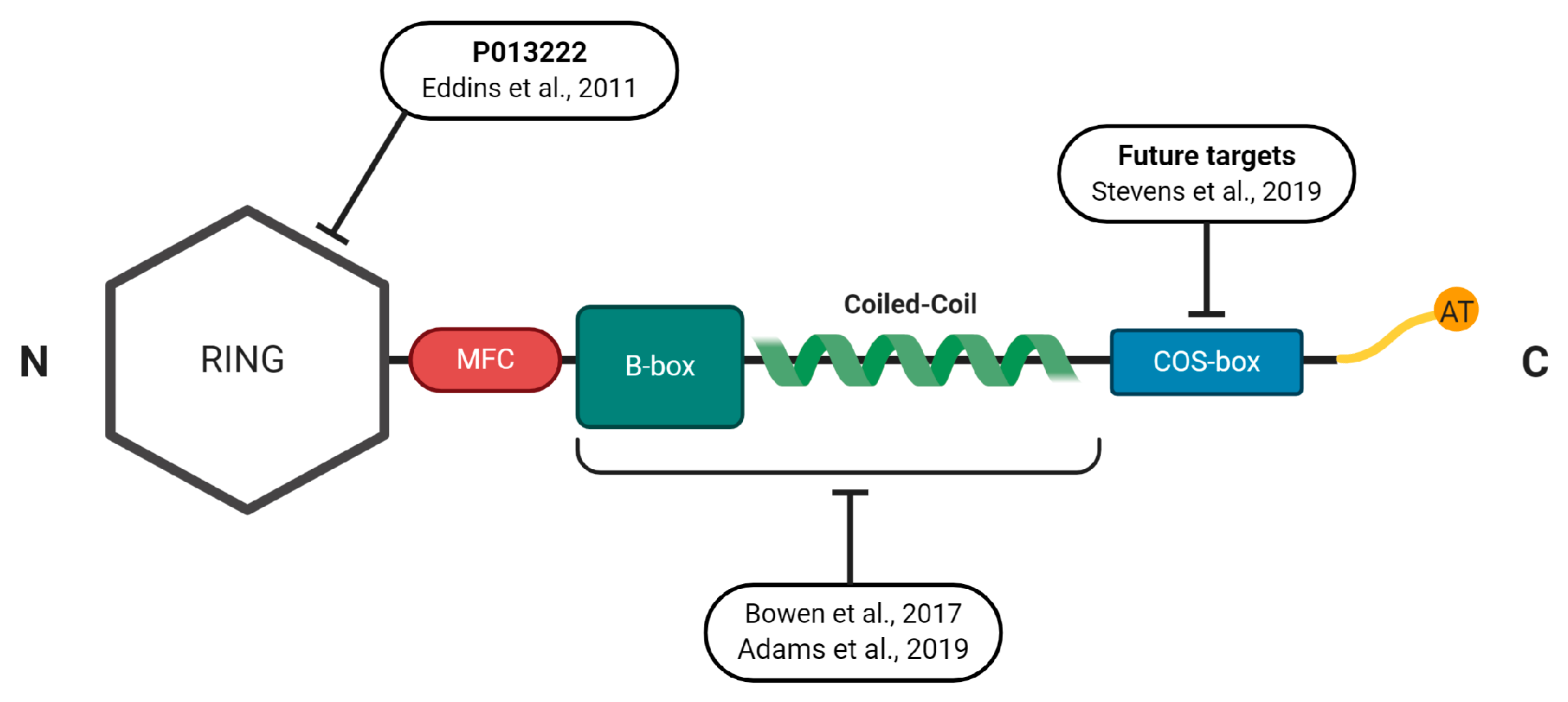
4.1. Structural Properties of the MuRFs
4.2. Developing Novel Small-Molecules to Inhibit MuRF1
4.3. Other Novel Approaches to Inhibit MuRF1 Function
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HOB | 5-hydroxybenzothiazolone |
| β2-AR | β2-adrenergic receptor |
| ActRIIB/A | activin receptors type IIB and IIA |
| Akt | protein kinase B |
| ATF4 | activating transcription factor 4 |
| BMP | TGF-β/myostatin/bone morphogenetic protein |
| C/EBP-δ | CCAAT-enhancer binding protein delta |
| CC | coiled coil |
| CHIP | C terminus of HSC70-interacting protein |
| COS-box | C-terminal subgroup one signature |
| Fbxo40 | F-box protein 40 |
| FoxO | Forkhead box protein O |
| GDF11 | growth and differentiation factor 11 |
| HMB | Beta-Hydroxy b-methylbutyrate |
| IGF1 | insulin-like growth gactor 1 |
| IkBa | kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| IkKb | inhibitor of nuclear factor kappa-B kinase subunit beta |
| IL-1 | Interleukin-1 |
| IL-6 | Interleukin-6 |
| MAFBx | muscle atrophy F-box |
| MFC | MuRF-family-specific motif |
| mTOR | mammalian target of rapamycin |
| MuRF1 | muscle-specific E3 ligase RING-finger protein 1 |
| MuRF2 | muscle-specific E3 ligase RING-finger protein 2 |
| MuRF3 | muscle-specific E3 ligase RING-finger protein 3 |
| MuRFs | muscle-specific RING finger proteins |
| MUSA1 | muscle ubiquitin ligase of SCF complex in atrophy-1 |
| NFkB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| PDH | pyruvate dehydrogenase |
| PROTACs | PROteolysis TArgeting Chimeras |
| RING | really interesting new gene |
| ROS | reactive oxygen species |
| Smad | mothers against decapentaplegic homolog |
| SMART | specific of muscle atrophy and regulated by transcription |
| SRF | serum response factor |
| Stat-3 | signal transducer and activator of transcription 3 |
| SUMO | small ubiquitin-like modifier |
| TGF-beta | transforming growth factor-beta |
| TNF alpha | tumor necrosis factor alpha |
| TRAF6 | tumor necrosis factor receptor (TNFR)-associated factor 6 |
| TRIM | TRIpartite motif |
| Trim 32 | tripartite motif-containing protein 32 |
| UPS | ubiquitin proteasome system |
References
- Frontera, W.R.; Ochala, J. Skeletal Muscle: A Brief Review of Structure and Function. Behav. Genet. 2015, 45, 183–195. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. DMM Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cachexia and sarcopenia: Mechanisms and potential targets for intervention. Curr. Opin. Pharmacol. 2015, 22, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Lynch, G.S.; Murphy, K.T.; Reid, M.B.; Zijdewind, I. Disease-Induced Skeletal Muscle Atrophy and Fatigue. Med. Sci. Sport Exerc. 2016, 48, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Gautel, M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat. Rev. Mol. Cell Biol. 2011, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2014, 14, 58–74. [Google Scholar] [CrossRef]
- Wang, X.H.; Mitch, W.E. Mechanisms of muscle wasting in chronic kidney disease. Nat. Rev. Nephrol. 2014, 10, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.A.V.; Al-Khalaf, M.; Megeney, L.A. The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet. Muscle 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pandurangan, M.; Hwang, I. The role of calpain in skeletal muscle. Anim. Cells Syst. 2012, 16, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Judge, A.R.; Bodine, S.C. CrossTalk opposing view: The dominant mechanism causing disuse muscle atrophy is proteolysis. J. Physiol. 2014, 592, 5345–5347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murton, A.J.; Constantin, D.; Greenhaff, P.L. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2008, 1782, 730–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, N.; Lee, K.K.; Jha, S. Targeting the Ubiquitin Proteasome System in Cancer. In Neoplasm; InTech: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Bartoli, M.; Richard, I. Calpains in muscle wasting. Int. J. Biochem. Cell Biol. 2005, 37, 2115–2133. [Google Scholar] [CrossRef]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigaré, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef]
- Bulatov, E.; Zagidullin, A.A.; Valiullina, A.; Sayarova, R.; Rizvanov, A.A. Small Molecule Modulators of RING-Type E3 Ligases: MDM and Cullin Families as Targets. Front. Pharmacol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Expressed During Muscle Atrophy. Structure 2001, 1–6. [Google Scholar]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [Green Version]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-related loss of muscle mass and function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 Ligase MuRF1 Degrades Myosin Heavy Chain Protein in Dexamethasone-Treated Skeletal Muscle. Cell. Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polge, C.; Heng, A.-E.; Jarzaguet, M.; Ventadour, S.; Claustre, A.; Combaret, L.; Bechet, D.; Matondo, M.; Uttenweiler, S.; Monsarrat, B.; et al. Muscle actin is polyubiquitinylated in vitro and in vivo and targeted for breakdown by the E3 ligase MuRF1. FASEB J. 2011, 25, 3790–3802. [Google Scholar] [CrossRef]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.K.; Gupta, S.K.; Bhatnagar, S.; Panguluri, S.K.; Darnay, B.G.; Choi, Y.; Kumar, A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 2010, 191, 1395–1411. [Google Scholar] [CrossRef] [Green Version]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-Assisted Selective Autophagy Is Essential for Muscle Maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Luo, L.; Eash, J.; Ibebunjo, C.; Glass, D.J. The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev. Cell. 2011, 21, 835–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.-H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Jokl, E.J.; Blanco, G. Disrupted autophagy undermines skeletal muscle adaptation and integrity. Mamm. Genome 2016, 27, 525–537. [Google Scholar] [CrossRef]
- Bilodeau, P.A.; Coyne, E.S.; Wing, S.S. The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation. Am. J. Physiol. Cell Physiol. 2016, 311, C392–C403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, J.J.; Michele, D.E.; Brooks, S.V. Inhibition of calpain prevents muscle weakness and disruption of sarcomere structure during hindlimb suspension. J. Appl. Physiol. 2010, 108, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supinski, G.S.; Wang, L.; Song, X.H.; Moylan, J.S.; Callahan, L.A. Muscle-specific calpastatin overexpression prevents diaphragm weakness in cecal ligation puncture-induced sepsis. J. Appl. Physiol. 2014, 117, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supinski, G.S.; Callahan, L.A. Caspase activation contributes to endotoxin-induced diaphragm weakness. J. Appl. Physiol. 2006, 100, 1770–1777. [Google Scholar] [CrossRef]
- Stratos, I.; Li, Z.; Rotter, R.; Herlyn, P.; Mittlmeier, T.; Vollmar, B. Inhibition of caspase mediated apoptosis restores muscle function after crush injury in rat skeletal muscle. Apoptosis 2012, 17, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Denis Alexander, H.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 1–28. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.-P. Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J. Cell Biochem. 2007, 100, 960–969. [Google Scholar] [CrossRef] [Green Version]
- Caron, A.Z.; Haroun, S.; Leblanc, E.; Trensz, F.; Guindi, C.; Amrani, A.; Grenier, G. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet. Disord. 2011, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Baos, S.; Prieto-Potin, I.; Román-Blas, J.A.; Sánchez-Pernaute, O.; Largo, R.; Herrero-Beaumont, G. Mediators and patterns of muscle loss in chronic systemic inflammation. Front. Physiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yakabe, M.; Ogawa, S.; Ota, H.; Iijima, K.; Eto, M.; Ouchi, Y.; Akishita, M. Inhibition of interleukin-6 decreases atrogene expression and ameliorates tail suspension-induced skeletal muscle atrophy. PLoS ONE 2018, 13, e20592. [Google Scholar] [CrossRef] [PubMed]
- Adams, V.; Mangner, N.; Gasch, A.; Krohne, C.; Gielen, S.; Hirner, S.; Thierse, H.-J.; Witt, C.C.; Linke, A.; Schuler, G.; et al. Induction of MuRF1 Is Essential for TNF-α-Induced Loss of Muscle Function in Mice. J. Mol. Biol. 2008, 384, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.-P.G.; Petersen, M.C.; Abudukadier, A.; Moreira, G.V.; Jurczak, M.J.; Friedman, G.; Haqq, C.M.; Petersen, K.F.; Shulman, G.I. Anti-myostatin antibody increases muscle mass and strength and improves insulin sensitivity in old mice. Proc. Natl. Acad. Sci USA 2016, 113, 2212–2217. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Coker, C.C.; Jirousek, M.R.; Zimmer, M.; Walter, G.A.; Sweeney, H.L. Disease-modifying effects of orally bioavailable NF-κB inhibitors in dystrophin-deficient muscle. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Zimmers, T.A.; Jiang, Y.; Wang, M.; Liang, T.W.; Rupert, J.E.; Au, E.; Marino, F.E.; Couch, M.E.; Koniaris, L.G. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res. Cardiol. 2017, 112, 48. [Google Scholar] [CrossRef]
- Jin, Q.; Qiao, C.; Li, J.; Xiao, B.; Li, J.; Xiao, X. A GDF11/myostatin inhibitor, GDF11 propeptide-Fc, increases skeletal muscle mass and improves muscle strength in dystrophic mdx mice. Skelet. Muscle. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 Activation Links a C/EBPδ to Myostatin Pathway to Stimulate Loss of Muscle Mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef]
- Frost, R.A.; Nystrom, G.J.; Jefferson, L.S.; Lang, C.H. Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 292. [Google Scholar] [CrossRef] [Green Version]
- Tiao, G.; Fagan, J.; Roegner, V.; Lieberman, M.; Wang, J.J.; Fischer, J.E.; Hasselgren, P.O. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Investig. 1996, 97, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Fang, Q.; Xu, T.; Wu, C.; Xu, L.; Wang, L.; Yang, X.; Yu, S.; Zhang, Q.; Ding, F.; et al. Mechanistic role of reactive oxygen species and therapeutic potential of antioxidants in denervation-or fasting-induced skeletal muscle atrophy. Front. Physiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Hudson, M.B.; Nelson, W.; Talbert, E.E.; Min, K.; Szeto, H.H.; Kavazis, A.N.; Smuder, A.J. Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness*. Crit. Care Med. 2011, 39, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrigo, J.; Rivera, J.C.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Transforming growth factor type beta (TGF-β) requires reactive oxygen species to induce skeletal muscle atrophy. Cell Signal 2016, 28, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Rinker, L.; Peng, J.; Chilian, W.M. Reactive Oxygen Species: The Good and the Bad. In Reactive Oxygen Species (ROS) in Living Cells; InTech: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Rothwarf, D.M.; Karin, M. The NF-kappa B activation pathway: A paradigm in information transfer from membrane to nucleus. Sci. STKE 1999, 1999. [Google Scholar] [CrossRef]
- Belova, S.P.; Shenkman, B.S.; Kostrominova, T.Y.; Nemirovskaya, T.L. Paradoxical effect of IKKβ inhibition on the expression of E3 ubiquitin ligases and unloading-induced skeletal muscle atrophy. Physiol. Rep. 2017, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Langenbacher, A.D.; Huang, J.; Wang, K.; Otto, G.W.; Geisler, R.; Wang, Y.; Chen, J.-N. The calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. eLife 2017, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jamart, C.; Raymackers, J.M.; An, G.L.; Deldicque, L.; Francaux, M. Prevention of muscle disuse atrophy by MG132 proteasome inhibitor. Muscle Nerve 2011, 43, 708–715. [Google Scholar] [CrossRef]
- Baptista, I.L.; Silvestre, J.G.; Silva, W.J.; Labeit, S.; Moriscot, A.S. FoxO3a suppression and VPS34 activity are essential to anti-atrophic effects of leucine in skeletal muscle. Cell Tissue Res. 2017, 369, 381–394. [Google Scholar] [CrossRef]
- Baptista, I.L.; Leal, M.L.; Artioli, G.G.; Aoki, M.S.; Fiamoncini, J.; Turri, A.O.; Curi, R.; Miyabara, E.H.; Moriscot, A. Leucine attenuates skeletal muscle wasting via inhibition of ubiquitin ligases. Muscle Nerve 2010, 41, 800–808. [Google Scholar] [CrossRef]
- Smith, H.J.; Mukerji, P.; Tisdale, M.J. Attenuation of proteasome-induced proteolysis in skeletal muscle by β-hydroxy-β-methylbutyrate in cancer-induced muscle loss. Cancer Res. 2005, 65, 277–283. [Google Scholar]
- Hao, Y.; Jackson, J.R.; Wang, Y.; Edens, N.; Pereira, S.L.; Alway, S.E. β-Hydroxy-β-methylbutyrate reduces myonuclear apoptosis during recovery from hind limb suspension-induced muscle fiber atrophy in aged rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.K.; Chung, K.W.; Choi, Y.J.; Park, M.H.; Jang, E.J.; Park, C.H.; Yoon, C.; Kim, N.D.; Kim, M.K.; Chung, H.Y. β-hydroxy β-methylbutyrate improves dexamethasone-induced muscle atrophy by modulating the muscle degradation pathway in SD rat. PLoS ONE 2014, 9, e102947. [Google Scholar] [CrossRef]
- Holeček, M. Beta-hydroxy-beta-methylbutyrate supplementation and skeletal muscle in healthy and muscle-wasting conditions. J. Cachexia Sarcopenia Muscle 2017, 8, 529–541. [Google Scholar] [CrossRef]
- Chen, L.; Chen, L.; Wan, L.; Huo, Y.; Huang, J.; Li, J.; Lu, J.; Xin, B.; Yang, Q.; Guo, C. Matrine improves skeletal muscle atrophy by inhibiting E3 ubiquitin ligases and activating the Akt/mTOR/FoxO3α signaling pathway in C2C12 myotubes and mice. Oncol. Rep. 2019, 42, 479–494. [Google Scholar] [CrossRef]
- Lynch, G.S.; Ryall, J.G. Role of β-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008, 88, 729–767. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Degracia, A.; Busquets, S.; Argilés, J.M.; Bargalló-Gispert, N.; López-Soriano, F.J.; Barreiro, E. Effects of the beta 2 agonist formoterol on atrophy signaling, autophagy, and muscle phenotype in respiratory and limb muscles of rats with cancer-induced cachexia. Biochimie 2018, 149, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Koziczak-Holbro, M.; Rigel, D.F.; Dumotier, B.; Sykes, D.A.; Tsao, J.; Nguyen, N.-H.; Bosch, J.; Jourdain, M.; Flotte, L.; Adachi, Y.; et al. Pharmacological characterization of a novel 5-hydroxybenzothiazolone-derived b2-adrenoceptor agonist with functional selectivity for anabolic effects on skeletal muscle resulting in a wider cardiovascular safety window in preclinical studiess. J. Pharmacol. Exp. Ther. 2019, 369, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Pötsch, M.S.; Ishida, J.; Palus, S.; Tschirner, A.; Von Haehling, S.; Doehner, W.; Anker, S.D.; Springer, J. MT-102 prevents tissue wasting and improves survival in a rat model of severe cancer cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 594–605. [Google Scholar] [CrossRef]
- Pötsch, M.S.; Tschirner, A.; Palus, S.; Von Haehling, S.; Doehner, W.; Beadle, J.; Coats, A.J.S.; Anker, S.D.; Springer, J. The anabolic catabolic transforming agent (ACTA) espindolol increases muscle mass and decreases fat mass in old rats. J. Cachexia Sarcopenia Muscle 2014, 5, 149–158. [Google Scholar] [CrossRef]
- Penna, F.; Bonetto, A.; Aversa, Z.; Minero, V.G.; Fanelli, F.R.; Costelli, P.; Muscaritoli, M. Effect of the specific proteasome inhibitor bortezomib on cancer-related muscle wasting. J. Cachexia Sarcopenia Muscle 2016, 345–354. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Beehler, B.C.; Sleph, P.G.; Benmassaoud, L.; Grover, G.J. Reduction of Skeletal Muscle Atrophy by a Proteasome Inhibitor in a Rat Model of Denervation. Exp. Biol. Med. 2006, 231, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Van Hees, H.W.H.; Li, Y.-P.; Ottenheijm, C.A.C.; Jin, B.; Pigmans, C.J.C.; Linkels, M.; Dekhuijzen, P.N.R.; Heunks, L.M.A. Proteasome inhibition improves diaphragm function in congestive heart failure rats Hieronymus. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294. [Google Scholar] [CrossRef]
- Agten, A.; Maes, K.; Thomas, D.; Cielen, N.; Van Hees, H.W.; Dekhuijzen, R.P.; Decramer, M.; Gayan-Ramirez, G. Bortezomib partially protects the rat diaphragm from ventilator-induced diaphragm dysfunction. Crit. Care Med. 2012, 40, 2449–2455. [Google Scholar] [CrossRef]
- Smuder, A.J.; Nelson, W.B.; Hudson, M.B.; Kavazis, A.N.; Powers, S.K. Inhibition of the ubiquitin-proteasome pathway does not protect against ventilator-induced accelerated proteolysis or atrophy in the diaphragm. Anesthesiology 2014, 121, 115–126. [Google Scholar] [CrossRef] [Green Version]
- Hungria, V.; Crusoé, E.D.Q.; Bittencourt, R.I.; Maiolino, A.; Magalhães, R.J.P.; Sobrinho, J.D.N.; Pinto, J.V.; Fortes, R.C.; Moreira, E.D.S.; Tanaka, P.Y. New Proteasome Inhibitors in the Treatment of Multiple Myeloma. Hematol. Transfus. Cell Ther. 2019, 41, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, C.; Peng, X.; Kang, Q.; Deng, D.; Zhang, L.; Zheng, Y.; Wang, C.; Qiao, Z.; Guo, D.; et al. Combined Treatment of Carfilzomib and Z-VAD-Fmk Inhibits Skeletal Proteolysis and Apoptosis and Ameliorates Cancer Cachexia. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, H.; Kou, Y.; Li, R.; Zheng, Y.; Wang, Q.; Zhou, X.; Jin, L. MG132-mediated inhibition of the ubiquitin-proteasome pathway ameliorates cancer cachexia. J. Cancer Res. Clin. Oncol. 2013, 139, 1105–1115. [Google Scholar] [CrossRef]
- Adams, V.; Bowen, T.S.; Werner, S.; Barthel, P.; Amberger, C.; Konzer, A.; Graumann, J.; Sehr, P.; Lewis, J.; Provaznik, J.; et al. Small-molecule-mediated chemical knock-down of MuRF1/MuRF2 and attenuation of diaphragm dysfunction in chronic heart failure. J. Cachexia Sarcopenia Muscle 2019, 10, 1102–1115. [Google Scholar] [CrossRef]
- Guglielmi, V.; Nowis, D.; Tinelli, M.; Malatesta, M.; Paoli, L.; Marini, M.; Manganotti, P.; Sadowski, R.; Wilczyński, G.M.; Meneghini, V.; et al. Bortezomib-induced muscle toxicity in multiple myeloma. J. Neuropathol. Exp. Neurol. 2017, 76, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C. Drugging the undruggable: Targeting challenging E3 ligases for personalized medicine. Future Med. Chem. 2017, 9, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Castillero, E.; Alamdari, N.; Lecker, S.H.; Hasselgren, P.O. Suppression of atrogin-1 and MuRF1 prevents dexamethasone-induced atrophy of cultured myotubes. Metabolism 2013, 62, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Lange, S.; Sheikh, F. Breaking down protein degradation mechanisms in cardiac muscle. Trends Mol. Med. 2013, 19, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Z.; De La Torre, R.; Barling, A.; Tsujikawa, T.; Hornick, N.; Hanifin, J.; Simpson, E.; Wang, Y.; Swanzey, E.; et al. Trim32 Deficiency Enhances Th2 Immunity and Predisposes to Features of Atopic Dermatitis. J. Investig. Dermatol. 2017, 137, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Luo, W.; Zhang, Y.; Yang, R.; Li, X.; Guo, Y.; Zhang, C.; Yang, R.; Gao, W.-Q. Trim32 suppresses cerebellar development and tumorigenesis by degrading Gli1/sonic hedgehog signaling. Cell Death Differ. 2020, 27, 1286–1299. [Google Scholar] [CrossRef]
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.-L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Pizon, V.; Iakovenko, A.; Van Der Ven, P.F.M.; Kelly, R.; Fatu, C.; Fürst, D.O.; Karsenti, E.; Gautel, M. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J. Cell Sci. 2002, 115, 4469–4482. [Google Scholar] [CrossRef] [Green Version]
- Mayans, O.; Labeit, S. MuRFs Specialized Members of the TRIM/RBCC Family with Roles in the Regulation of the Trophic State of Muscle and Its Metabolism; Landes Bioscience: Austin, TX, USA; Springer: Berlin, Germany, 2012; pp. 119–130. [Google Scholar]
- Spencer, J.A.; Eliazer, S.; Ilaria, R.L.; Richardson, J.A.; Olson, E.N. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING-finger protein. J. Cell Biol. 2000, 150, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Esposito, D.; Koliopoulos, M.G.; Rittinger, K. Structural determinants of TRIM protein function. Biochem. Soc. Trans. 2017, 45, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Franke, B.; Skorupka, K.A.; Cafiso, D.S.; Pornillos, O.; Mayans, O.; Norman, D.G. Exploration of the TRIM Fold of MuRF1 Using EPR Reveals a Canonical Antiparallel Structure and Extended COS-Box. J. Mol. Biol. 2019, 431, 2900–2909. [Google Scholar] [CrossRef]
- Franke, B.; Gasch, A.; Rodriguez, D.; Chami, M.; Khan, M.M.; Rudolf, R.; Bibby, J.; Hanashima, A.; Bogomolovas, J.; Von Castelmur, E.; et al. Molecular basis for the fold organization and sarcomeric targeting of the muscle atrogin muRF1. Open Biol. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrosek, M.; Meier, S.; Ucurum-Fotiadis, Z.; Von Castelmur, E.; Hedbom, E.; Lustig, A.; Grzesiek, S.; Labeit, D.; Labeit, S.; Mayans, O. Structural Analysis of B-Box 2 from MuRF1: Identification of a Novel Self-Association Pattern in a RING-like Fold. Biochemistry 2008, 47, 10722–10730. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.G.; Baptista, I.L.; Carlassara, E.O.C.; Moriscot, A.S.; Aoki, M.S.; Miyabara, E.H. Leucine supplementation improves skeletal muscle regeneration after cryolesion in rats. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granzier, H.L.; Labeit, S. The Giant Protein Titin: A Major Player in Myocardial Mechanics, Signaling, and Disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, T.S.; Adams, V.; Werner, S.; Fischer, T.; Vinke, P.; Brogger, M.N.; Mangner, N.; Linke, A.; Sehr, P.; Lewis, J.; et al. Small-molecule inhibition of MuRF1 attenuates skeletal muscle atrophy and dysfunction in cardiac cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Mankoo, B.; Gautel, M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J. Muscle Res. Cell Motil. 2012, 33, 107–122. [Google Scholar] [CrossRef] [Green Version]
- Lodka, D.; Pahuja, A.; Geers-Knörr, C.; Scheibe, R.J.; Nowak, M.; Hamati, J.; Köhncke, C.; Purfürst, B.; Kanashova, T.; Schmidt, S.; et al. Muscle RING-finger 2 and 3 maintain striated-muscle structure and function. J. Cachexia Sarcopenia Muscle 2016, 7, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Mattox, T.A.; Young, M.E.; Rubel, C.E.; Spaniel, C.; Rodriguez, J.E.; Grevengoed, T.J.; Gautel, M.; Xu, Z.; Anderson, E.J.; Willis, M.S. MuRF1 activity is present in cardiac mitochondria and regulates reactive oxygen species production in vivo. J. Bioenerg. Biomembr. 2014, 46, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef]
- Nguyen, T.; Bowen, T.S.; Augstein, A.; Schauer, A.; Gasch, A.; Linke, A.; Labeit, S.; Adams, V. Expression of MuRF1 or MuRF2 is essential for the induction of skeletal muscle atrophy and dysfunction in a murine pulmonary hypertension model. Skelet. Muscle. 2020, 10, 12. [Google Scholar] [CrossRef]
- Olive, M.; Abdul-Hussein, S.; Oldfors, A.; González-Costello, J.; Van Der Ven, P.F.; Fürst, D.O.; González, L.; Moreno, D.; Torrejón-Escribano, B.; Alió, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.-S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Investig. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Fielitz, J.; Van Rooij, E.; Spencer, J.A.; Shelton, J.M.; Latif, S.; Van Der Nagel, R.; Bezprozvannaya, S.; De Windt, L.; Richardson, J.A.; Bassel-Duby, R.; et al. Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc. Natl. Acad. Sci. USA 2007, 104, 4377–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, M.T.; He, J.; Sullivan, J.; Grevengoed, T.J.; Schisler, J.; Han, Y.; Hill, J.A.; Yates, C.; Stansfield, E.W.; Mapanga, R.F.; et al. Muscle ring finger-3 protects against diabetic cardiomyopathy induced by a high fat diet. BMC Endocr. Disord. 2015, 15, e36. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, M.J.; Nicholson, B.; Kessler, B.M. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev. Mol. Med. 2011, 13, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Witt, C.C.; Witt, S.H.; Lerche, S.; Labeit, D.; Back, W.; Labeit, S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008, 27, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Wexler, B.C. Alloxan-induced diabetes in young vs old sprague-dawley rats. Exp. Gerontol. 1981, 16, 47–58. [Google Scholar] [CrossRef]
- Eddins, M.J.; Marblestone, J.G.; Kumar, K.G.S.; Leach, C.A.; Sterner, D.E.; Mattern, M.R.; Nicholson, B. Targeting the Ubiquitin E3 Ligase MuRF1 to Inhibit Muscle Atrophy. Cell Biochem. Biophys. 2011, 60, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef]
- Willis, M.S.; Wadosky, K.; Rodriguez, J.E.; Schisler, J.; Lockyer, P.; Hilliard, E.G.; Glass, D.J.; Patterson, C. Muscle ring finger 1 and muscle ring finger 2 are necessary but functionally redundant during developmental cardiac growth and regulate E2F1-mediated gene expression in vivo. Cell Biochem. Funct. 2014, 32, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirner, S.; Krohne, C.; Schuster, A.; Hoffmann, S.; Witt, S.; Erber, R.; Sticht, C.; Gasch, A.; Labeit, S.; Labeit, D. MuRF1-dependent Regulation of Systemic Carbohydrate Metabolism as Revealed from Transgenic Mouse Studies. J. Mol. Biol. 2008, 379, 666–677. [Google Scholar] [CrossRef]
- Crossland, H.; Skirrow, S.; Puthucheary, Z.A.; Constantin-Teodosiu, D.; Greenhaff, P.L. The impact of immobilisation and inflammation on the regulation of muscle mass and insulin resistance: Different routes to similar end-points. J. Physiol. 2019, 597, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Silvestre, J.; Baptista, I.L.; Silva, W.; Cruz, A.; Silva, M.; Miyabara, E.H.; Labeit, S.; Moriscot, A. The e3 ligase murf2 plays a key role in the functional capacity of skeletal muscle fibroblasts. Braz. J. Med. Biol. Res. 2019, 52, 1–10. [Google Scholar] [CrossRef]
- Heras, G.; Namuduri, A.V.; Traini, L.; Shevchenko, G.; Falk, A.; Lind, S.B.; Jia, M.; Tian, G.; Gastaldello, S.; Mi, J. Muscle RING-finger protein-1 (MuRF1) functions and cellular localization are regulated by SUMO1 post-translational modification. J. Mol. Cell Biol. 2019, 11, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Johnson, J.A.; Su, H. Ubiquitin and Ubiquitin-like Proteins in Cardiac Disease and Protection. Curr. Drug Targets 2018, 19, 989–1002. [Google Scholar] [CrossRef]
- Nowak, M.; Suenkel, B.; Porras, P.; Migotti, R.; Schmidt, F.; Kny, M.; Zhu, X.; Wanker, E.; Dittmar, G.; Fielitz, J.; et al. DCAF8, a Novel MuRF1 Interaction Partner, Promotes Muscle Atrophy. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.; Wang, S.; Jaime-Figueroa, S.; Harbin, A.; Wang, J.; Hamman, B.D.; Crews, C.M. Differential PROTAC Substrate Specificity Dictated by Orientation of Recruited E3 Ligase. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. Protacs: Great Opportunities for Academia and Industry. Signal Transduct. Targeted Ther. 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Ebert, S.M.; Dierdorff, J.M.; Meyerholz, D.K.; Bullard, S.A.; Al-Zougbi, A.; Delau, A.D.; Tomcheck, K.C.; Skopec, Z.P.; Marcotte, G.R.; Bodine, S.C.; et al. An investigation of p53 in skeletal muscle aging. J. Appl. Physiol. 2019, 127, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Mangner, N.; Bowen, T.S.; Werner, S.; Fischer, T.; Kullnick, Y.; Oberbach, A.; Linke, A.; Steil, L.; Schuler, G.; Adams, V. Exercise Training Prevents Diaphragm Contractile Dysfunction in Heart Failure. Med. Sci. Sport Exerc. 2016, 48, 2118–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gielen, S.; Sandri, M.; Kozarez, I.; Kratzsch, J.; Teupser, D.; Thiery, J.; Erbs, S.; Mangner, N.; Lenk, K.; Hambrecht, R.; et al. Exercise Training Attenuates MuRF-1 Expression in the Skeletal Muscle of Patients With Chronic Heart Failure Independent of Age: The Randomized Leipzig Exercise Intervention in Chronic Heart Failure and Aging Catabolism Study. Circulation 2012, 125, 2716–2727. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scalabrin, M.; Adams, V.; Labeit, S.; Bowen, T.S. Emerging Strategies Targeting Catabolic Muscle Stress Relief. Int. J. Mol. Sci. 2020, 21, 4681. https://doi.org/10.3390/ijms21134681
Scalabrin M, Adams V, Labeit S, Bowen TS. Emerging Strategies Targeting Catabolic Muscle Stress Relief. International Journal of Molecular Sciences. 2020; 21(13):4681. https://doi.org/10.3390/ijms21134681
Chicago/Turabian StyleScalabrin, Mattia, Volker Adams, Siegfried Labeit, and T. Scott Bowen. 2020. "Emerging Strategies Targeting Catabolic Muscle Stress Relief" International Journal of Molecular Sciences 21, no. 13: 4681. https://doi.org/10.3390/ijms21134681
APA StyleScalabrin, M., Adams, V., Labeit, S., & Bowen, T. S. (2020). Emerging Strategies Targeting Catabolic Muscle Stress Relief. International Journal of Molecular Sciences, 21(13), 4681. https://doi.org/10.3390/ijms21134681