Wnt/β-Catenin Signaling in Oral Carcinogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
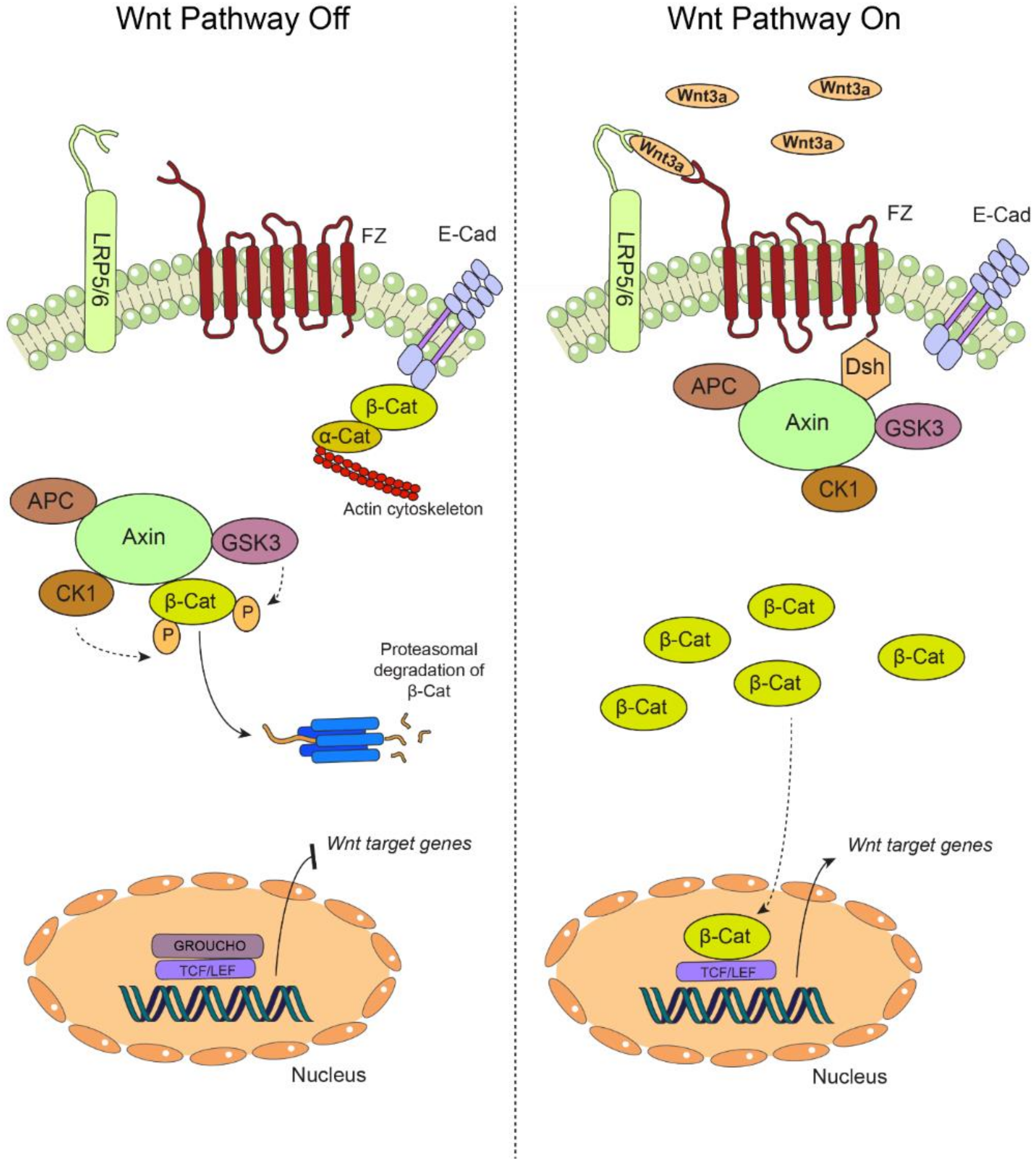
2. Canonical Wnt Pathway
3. Altered Wnt/β-Catenin Signaling in Oral Carcinogenesis
3.1. Oral Carcinogenesis
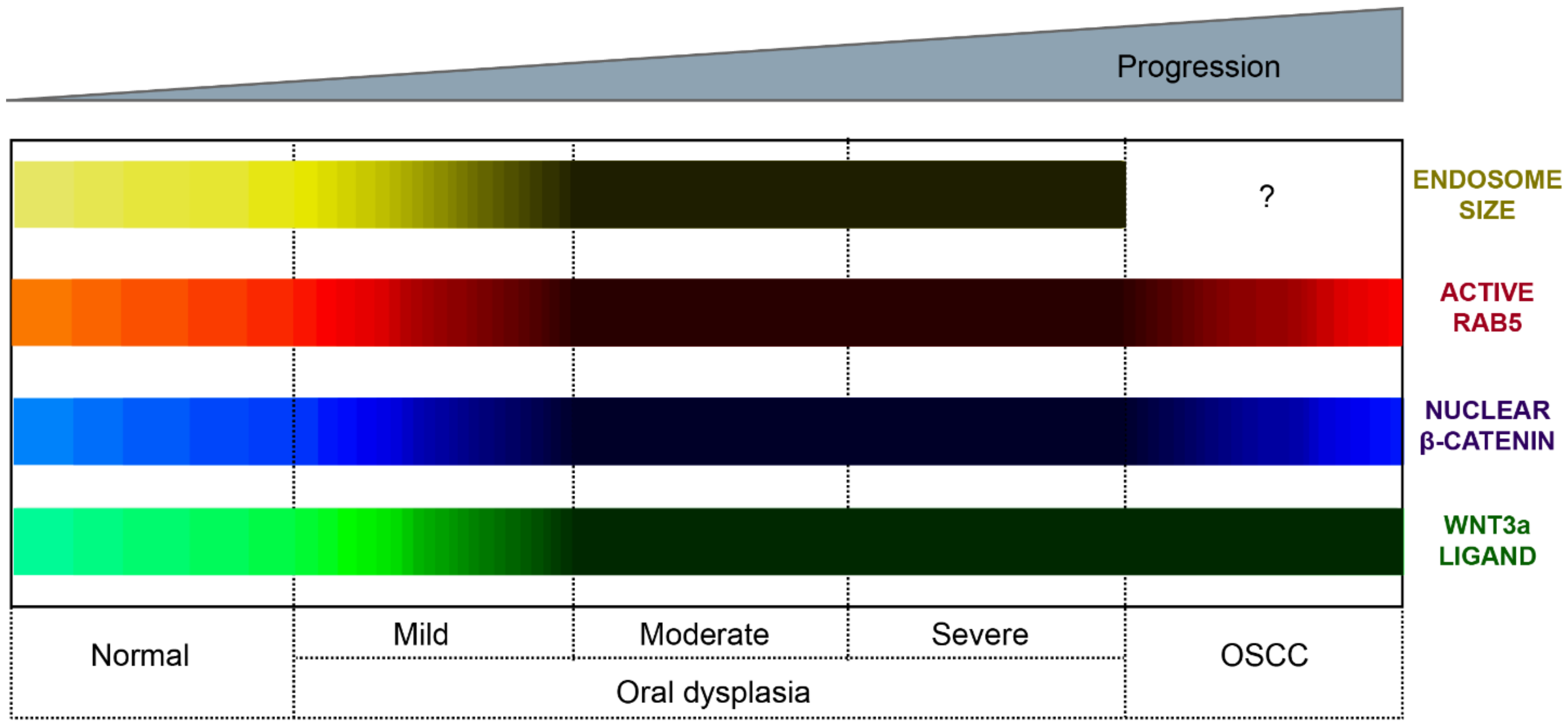
3.2. Wnt/β-Catenin Signaling in Oral Dysplasia
3.3. Wnt/β-Catenin Signaling in Oral Cancer
3.4. Wnt Inhibitors in Oral Carcinogenesis
4. Mechanisms Involved in the Aberrant Activation of β-Catenin in Oral Cancer
4.1. Endosomal Trafficking and Wnt/β-Catenin Signaling
4.2. Deregulated Endocytosis in Cancer
4.3. Endosomal Sequestration of the Destruction Complex in Oral Dysplasia
5. Therapeutic Approaches Based on Targeting Wnt Secretion
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Haddad, R.I.; Shin, D.M. Recent advances in head and neck cancer. N. Engl. J. Med. 2008, 359, 1143–1154. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Neville, B.W.; Day, T.A. Oral cancer and precancerous lesions. CA. Cancer J. Clin. 2002, 52, 195–215. [Google Scholar] [CrossRef]
- Polanska, H.; Raudenska, M.; Gumulec, J.; Sztalmachova, M.; Adam, V.; Kizek, R.; Masarik, M. Clinical significance of head and neck squamous cell cancer biomarkers. Oral Oncol. 2014, 50, 168–177. [Google Scholar] [CrossRef]
- Genden, E.M.; Ferlito, A.; Silver, C.E.; Takes, R.P.; Suarez, C.; Owen, R.P.; Haigentz, M.; Stoeckli, S.J.; Shaha, A.R.; Rapidis, A.D.; et al. Contemporary management of cancer of the oral cavity. Eur. Arch. Otorhinolaryngol. 2010, 267, 1001–1017. [Google Scholar] [CrossRef] [Green Version]
- Dost, F.; Le Cao, K.; Ford, P.J.; Ades, C.; Farah, C.S. Malignant transformation of oral epithelial dysplasia: A real-world evaluation of histopathologic grading. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2014, 117, 343–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnakulasuriya, S.; Reibel, J.; Bouquot, J.; Dabelsteen, E. Oral epithelial dysplasia classification systems: Predictive value, utility, weaknesses and scope for improvement. J. Oral. Pathol. Med. 2008, 37, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Awadallah, M.; Idle, M.; Patel, K.; Kademani, D. Management update of potentially premalignant oral epithelial lesions. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2018, 125, 628–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietel, M.; Johrens, K.; Laffert, M.V.; Hummel, M.; Blaker, H.; Pfitzner, B.M.; Lehmann, A.; Denkert, C.; Darb-Esfahani, S.; Lenze, D.; et al. A 2015 update on predictive molecular pathology and its role in targeted cancer therapy: A review focussing on clinical relevance. Cancer Gene Ther. 2015, 22, 417–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgakopoulou, E.A.; Stebbing, J.; Scully, C. Targeted cancer therapies. J. Am. Dent. Assoc. 2018, 149, 100–111. [Google Scholar] [CrossRef]
- Reyes, M.; Pena-Oyarzun, D.; Maturana, A.; Torres, V.A. Nuclear localization of beta-catenin and expression of target genes are associated with increased Wnt secretion in oral dysplasia. Oral. Oncol. 2019, 94, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Sha, J.; Kanno, T. The role of carcinogenesis-related biomarkers in the wnt pathway and their effects on Epithelial–Mesenchymal Transition (EMT) in oral squamous cell carcinoma. Cancers 2020, 12, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, F.A.P.; Noguti, J.; Oshima, C.T.F.; Ribeiro, D.A. Effective targeting of the epidermal growth factor receptor (EGFR) for treating oral cancer: A promising approach. Anticancer Res. 2014, 34, 1547–1552. [Google Scholar] [PubMed]
- Reyes, M.; Pena-Oyarzun, D.; Silva, P.; Venegas, S.; Criollo, A.; Torres, V.A. Nuclear accumulation of beta-catenin is associated with endosomal sequestration of the destruction complex and increased activation of Rab5 in oral dysplasia. Faseb J. 2020, 34, 4009–4025. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Ito, S.; Wada, N.; Deguchi, H.; Hata, T.; Hosoda, M.; Nohno, T. Nuclear localization of beta-catenin involved in precancerous change in oral leukoplakia. Mol. Cancer 2007, 6, 62. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Okazaki, Y.; Tonogi, M.; Tanaka, Y.; Yamane, G.-Y. Expression of beta-catenin in rat oral epithelial dysplasia induced by 4-nitroquinoline 1-oxide. Oral Oncol. 2002, 38, 772–778. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Boil. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R. Wnt signaling. Cold Spring Harb. Perspect. Boil. 2012, 4, a011163. [Google Scholar] [CrossRef]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.-J. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef]
- Santoro, A.; Pannone, G.; Papagerakis, S.; McGuff, H.S.; Cafarelli, B.; Lepore, S.; De Maria, S.; Rubini, C.; Mattoni, M.; Staibano, S.; et al. Beta-catenin and epithelial tumors: A study based on 374 oropharyngeal cancers. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, P.; Zhang, B.; Xi, G.-M.; Wu, Y.; Liu, H.-B.; Liu, Y.-F.; Xu, W.-J.; Zhu, Q.-Q.; Cai, F.; Zhou, Z.-J.; et al. PRC1 contributes to tumorigenesis of lung adenocarcinoma in association with the Wnt/β-catenin signaling pathway. Mol. Cancer 2017, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Khalil, S.; Tan, G.A.; Giri, D.D.; Zhou, X.K.; Howe, L. Activation status of Wnt/ß-catenin signaling in normal and neoplastic breast tissues: Relationship to HER2/NEU expression in human and mouse. PLoS ONE 2012, 7, e33421. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, G.; Devaraj, H. Aberrant expression of beta-catenin and its association with DeltaNp63, Notch-1, and clinicopathological factors in oral squamous cell carcinoma. Clin. Oral. Investig. 2012, 16, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, M.; Andiappan, M.; Wahab, A.; Muthusekhar, M.R.; Balakrishnan, A.; Shanmugam, S. Canonical Wnt pathway gene expression and their clinical correlation in oral squamous cell carcinoma. Indian J. Dent. Res. 2018, 29, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of beta-catenin and E-cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Rojas-Alcayaga, G.; Maturana, A.; Aitken, J.P.; Rojas, C.; Ortega, A.V. Increased nuclear beta-catenin expression in oral potentially malignant lesions: A marker of epithelial dysplasia. Med. Oral. Patol. Oral. Cir. Bucal. 2015, 20, e540. [Google Scholar] [CrossRef]
- Iwai, S.; Katagiri, W.; Kong, C.; Amekawa, S.; Nakazawa, M.; Yura, Y. Mutations of the APC, beta-catenin, and axin 1 genes and cytoplasmic accumulation of beta-catenin in oral squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2005, 131, 773–782. [Google Scholar] [CrossRef]
- Gasparoni, A.; Chaves, A.; Fonzi, L.; Johnson, G.K.; Schneider, G.B.; Squier, C.A. Subcellular localization of beta-catenin in malignant cell lines and squamous cell carcinomas of the oral cavity. J. Oral Pathol. Med. 2002, 31, 385–394. [Google Scholar] [CrossRef]
- Tsuchiya, R.; Yamamoto, G.; Nagoshi, Y.; Aida, T.; Irié, T.; Tachikawa, T. Expression of adenomatous polyposis coli (APC) in tumorigenesis of human oral squamous cell carcinoma. Oral Oncol. 2004, 40, 932–940. [Google Scholar] [CrossRef]
- Yeh, K.-T.; Chang, J.-G.; Lin, T.-H.; Wang, Y.-F.; Chang, J.-Y.; Shih, M.-C.; Lin, C.-C. Correlation between protein expression and epigenetic and mutation changes of Wnt pathway-related genes in oral cancer. Int. J. Oncol. 2003, 23, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Uraguchi, M.; Morikawa, M.; Shirakawa, M.; Sanada, K.; Imai, K. Activation of WNT family expression and signaling in squamous cell carcinomas of the oral cavity. J. Dent. Res. 2004, 83, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Odajima, T.; Sasaki, Y.; Tanaka, N.; Kato-Mori, Y.; Asanuma, H.; Ikeda, T.; Satoh, M.; Hiratsuka, H.; Tokino, T.; Sawada, N. Abnormal beta-catenin expression in oral cancer with no gene mutation: Correlation with expression of cyclin D1 and epidermal growth factor receptor, Ki-67 labeling index, and clinicopathological features. Hum. Pathol. 2005, 36, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Nusse, R. Wnt Proteins. Cold Spring Harb. Perspect. Boil. 2012, 4, a007864. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.-M.; Niehrs, C. Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb. Perspect. Boil. 2012, 5, a015081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, B.T.; He, X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb Perspect. Biol. 2012, 4, a007880. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Mikels, A.J.; Nusse, R. Wnts as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.; Nusse, R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J. Boil. Chem. 2006, 281, 22429–22433. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, R.G.; Arai, M.A.; Ishibashi, M. Natural compounds with Wnt signal modulating activity. Nat. Prod. Rep. 2015, 32, 1622–1628. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Nagse, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef]
- Boynton, R.F.; Blount, P.L.; Yin, J.; Brown, V.L.; Huang, Y.; Tong, Y.; McDaniel, T.; Newkirk, C.; Resau, J.H.; Raskind, W.H. Loss of heterozygosity involving the APC and MCC genetic loci occurs in the majority of human esophageal cancers. Proc. Natl. Acad. Sci. USA 1992, 89, 3385–3388. [Google Scholar] [CrossRef] [Green Version]
- Korinek, V.; Barker, N.; Morin, P.J.; Wichen, D.V.; Weger, R.D.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a β-catenin-tcf complex in apc−/−colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [Green Version]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef]
- Coste, A.D.L.; Romagnolo, B.; Billuart, P.; Renard, C.-A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the -catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef] [Green Version]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Phoon, Y.P.; Jin, X.; Chong, S.Y.S.; Ip, J.C.Y.; Wong, B.W.Y.; Lung, M.L. Wnt-C59 arrests stemness and suppresses growth of nasopharyngeal carcinoma in mice by inhibiting the Wnt pathway in the tumor microenvironment. Oncotarget 2015, 6, 14428–14439. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, L.; Melo, F.D.S.E.; Van Der Heijden, M.; Cameron, K.; De Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; I Smith, D.; et al. Mutational spectrum of β-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Fukuchi, M.; Miyazaki, T.; Masuda, N.; Kato, H.; Kuwano, H. Reduced expression of Axin correlates with tumour progression of oesophageal squamous cell carcinoma. Br. J. Cancer 2003, 88, 1734–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trumper, L.; Binder, C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef] [Green Version]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.-C. β-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef] [Green Version]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Rothberg, B.E.G.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-Catenin signaling controls metastasis in braf-activated pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Hanaki, H.; Yamamoto, H.; Sakane, H.; Matsumoto, S.; Ohdan, H.; Sato, A.; Kikuchi, A. An anti-wnt5a antibody suppresses metastasis of gastric cancer cells in vivo by inhibiting receptor-mediated endocytosis. Mol. Cancer Ther. 2012, 11, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.-C.; Lee, P.-T.; Wu, M.-H.; Huang, C.-C.; Ko, C.-Y.; Lee, Y.-C.; Lin, D.-Y.; Cheng, Y.-W.; Lee, K.-H. Distinct roles and differential expression levels of Wnt5a mRNA isoforms in colorectal cancer cells. PLoS ONE 2017, 12, e0181034. [Google Scholar] [CrossRef]
- Wang, T.; Liu, X.; Wang, J. Up-regulation of Wnt5a inhibits proliferation and migration of hepatocellular carcinoma cells. J. Cancer Res. Ther. 2019, 15, 904–908. [Google Scholar] [CrossRef]
- Jia, S.; Qu, T.; Feng, M.; Ji, K.; Li, Z.; Jiang, W.G.; Ji, J. Association of Wnt1-inducible signaling pathway protein-1 with the proliferation, migration and invasion in gastric cancer cells. Tumor Boil. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, K.; Wu, J.; Shi, J.; Xue, J.; Li, J.; Chen, J.; Zhu, Y.; Wei, J.; He, J.; et al. Wnt5a increases properties of lung cancer stem cells and resistance to cisplatin through activation of Wnt5a/PKC signaling pathway. Stem Cells Int. 2016, 2016, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tang, Z.; Gong, H.; Zhu, L.; Liu, X. Wnt5a promotes epithelial-to-mesenchymal transition and metastasis in non-small-cell lung cancer. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Whang, Y.M.; Jo, U.; Sung, J.S.; Ju, H.J.; Kim, H.K.; Park, K.H.; Lee, J.W.; Koh, I.S.; Kim, Y.H. Wnt5a is associated with cigarette smoke-related lung carcinogenesis via protein kinase C. PLoS ONE 2013, 8, e53012. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Tinsley, H.; Keeton, A.; Qu, Z.; Piazza, G.A.; Li, Y. Suppression of Wnt/β-catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 2008, 602, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Cheng, L.; Li, J.; Farah, E.; Lanman, N.A.; Pascuzzi, P.; Gupta, S.; Liu, X. Inhibition of the Wnt/β-catenin pathway overcomes resistance to enzalutamide in castration-resistant prostate cancer. Cancer Res. 2018, 78, 3147–3162. [Google Scholar] [CrossRef] [Green Version]
- Aldahl, J.; Mi, J.; Pineda, A.; Kim, W.K.; Olson, A.; Hooker, E.; He, Y.; Yu, E.-J.; Le, V.; Lee, D.-H.; et al. Aberrant activation of hepatocyte growth factor/MET signaling promotes β-catenin–mediated prostatic tumorigenesis. J. Boil. Chem. 2020, 295, 631–644. [Google Scholar] [CrossRef]
- Prgomet, Z.; Andersson, T.; Lindberg, P. Higher expression of WNT5A protein in oral squamous cell carcinoma compared with dysplasia and oral mucosa with a normal appearance. Eur. J. Oral Sci. 2017, 125, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Prgomet, Z.; Axelsson, L.; Lindberg, P.; Andersson, T. Migration and invasion of oral squamous carcinoma cells is promoted by WNT5A, a regulator of cancer progression. J. Oral Pathol. Med. 2014, 44, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Filho, P.A.; Letra, A.; Cramer, A.; Prasad, J.; Garlet, G.P.; Vieira, A.; Ferris, R.; Menezes, R. Insights from studies with oral cleft genes suggest associations between WNT-pathway genes and risk of oral cancer. J. Dent. Res. 2011, 90, 740–746. [Google Scholar] [CrossRef]
- Sogabe, Y.; Suzuki, H.; Toyota, M.; Ogi, K.; Imai, T.; Nojima, M.; Sasaki, Y.; Hiratsuka, H.; Tokino, T. Epigenetic inactivation of SFRP genes in oral squamous cell carcinoma. Int. J. Oncol. 2008, 32, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasser, W.; Flechtenmacher, C.; Holzinger, D.; Hofele, C.; Bosch, F.X. Aberrant expression of p53, p16INK4a and Ki-67 as basic biomarker for malignant progression of oral leukoplakias. J. Oral Pathol. Med. 2011, 40, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Trivedi, T.; Tankshali, R.; Goswami, J.; Shah, J.; Jetly, D.; Kobawala, T.; Patel, K.; Shukla, S.; Shah, P.; et al. Molecular alterations in oral carcinogenesis: Significant risk predictors in malignant transformation and tumor progression. Int. J. Boil. Markers 2007, 22, 132–143. [Google Scholar] [CrossRef]
- Ramakrishna, A.; Shreedhar, B.; Narayan, T.; Mohanty, L.; Shenoy, S.; Jamadar, S. Cyclin D1 an early biomarker in oral carcinogenesis. J. Oral Maxillofac. Pathol. 2013, 17, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Cordell, K.G.; Lee, J.S.; Worden, F.P.; Prince, M.E.; Tran, H.H.; Wolf, G.T.; Urba, S.G.; Chepeha, D.B.; Teknos, T.N.; et al. EGFR, p16, HPV titer, Bcl-xL and p53, sex, and smoking as indicators of response to therapy and survival in oropharyngeal cancer. J. Clin. Oncol. 2008, 26, 3128–3137. [Google Scholar] [CrossRef] [Green Version]
- Guan, G.; Bakr, M.M.; Firth, N.; Love, R.M. Expression of cyclin D1 correlates with p27KIP1 and regulates the degree of oral dysplasia and squamous cell carcinoma differentiation. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 126, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Ramasubramanian, A.; Ramani, P.; Sherlin, H.J.; Premkumar, P.; Natesan, A.; Thiruvengadam, C. Immunohistochemical evaluation of oral epithelial dysplasia using cyclin-D1, p27 and p63 expression as predictors of malignant transformation. J. Nat. Sci. Boil. Med. 2013, 4, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.G.; Trivedi, T.I.; Tankshali, R.A.; Goswami, J.V.; Jetly, D.H.; Shukla, S.N.; Shah, P.M.; Verma, R.J. Prognostic significance of molecular markers in oral squamous cell carcinoma: A multivariate analysis. Head Neck 2009, 31, 1544–1556. [Google Scholar] [CrossRef]
- Edwards, P.C. The natural history of oral epithelial dysplasia: Perspective on Dost et al. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 263–266. [Google Scholar] [CrossRef]
- Speight, P.M. Update on oral epithelial dysplasia and progression to cancer. Head Neck Pathol. 2007, 1, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Van Der Waal, I. Oral potentially malignant disorders: Is malignant transformation predictable and preventable? Med. Oral Patol. Oral Cir. Bucal 2014, 19, e386–e390. [Google Scholar] [CrossRef] [PubMed]
- Cervigne, N.K.; Machado, J.; Goswami, R.; Sadikovic, B.; Bradley, G.; Perez-Ordonez, B.; Galloni, N.N.; Gilbert, R.; Gullane, P.; Irish, J.C.; et al. Recurrent genomic alterations in sequential progressive leukoplakia and oral cancer: Drivers of oral tumorigenesis? Hum. Mol. Genet. 2014, 23, 2618–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Tabor, M.P.; Kummer, J.A.; Leemans, C.R.; Brakenhoff, R.H. A genetic explanation of Slaughter’s concept of field cancerization: Evidence and clinical implications. Cancer Res. 2003, 63, 1727–1730. [Google Scholar]
- Kumar, K.U.G.; Sathiasekar, A.C.; Mathew, D.G.; Lal, M.S.J.; Prakash, A.A.A. Oral field cancerization and its clinical implications in the management in potentially malignant disorders. J. Pharm. Bioallied Sci. 2017, 9, S23–S25. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, B.J.; Tabor, M.P.; Leemans, C.R.; Van Der Waal, I.; Snow, G.B.; Brakenhoff, R.H. Second primary tumors and field cancerization in oral and oropharyngeal cancer: Molecular techniques provide new insights and definitions. Head Neck 2002, 24, 198–206. [Google Scholar] [CrossRef]
- Francis, G.; Kumar, U.D.; Nalinakumari, K.; Jayasree, K.; Kannan, S. Accumulation of inactive p53 protein in oral squamous cell carcinoma: Stabilization by protein interaction. Eur. J. Oral Sci. 2013, 121, 21–28. [Google Scholar] [CrossRef]
- Tanaka, T.; Tanaka, M.; Tanaka, T. Oral carcinogenesis and oral cancer chemoprevention: A review. Pathol. Res. Int. 2011, 2011, 1–10. [Google Scholar] [CrossRef]
- Bodhade, A.S.; Dive, A.M. Chemoprevention of premalignant and malignant lesions of oral cavity: Recent trends. Eur. J. Dent. 2013, 7, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Warnakulasuriya, S.; Johnson, N.W.; Van Der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J. Oral Pathol. Med. 2007, 36, 575–580. [Google Scholar] [CrossRef]
- Fleskens, S.; Slootweg, P.J. Grading systems in head and neck dysplasia: Their prognostic value, weaknesses and utility. Head Neck Oncol. 2009, 1, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Naggar, A.K.; Chan, J.K.; Takata, T.; Grandis, J.R.; Slootweg, P.J. The fourth edition of the head and neck World Health Organization blue book: Editors’ perspectives. Hum. Pathol. 2017, 66, 10–12. [Google Scholar] [CrossRef]
- Nag, R.; Das, R.K. Analysis of images for detection of oral epithelial dysplasia: A review. Oral Oncol. 2018, 78, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Reichart, P.; Philipsen, H.P. Oral erythroplakia—A review. Oral Oncol. 2005, 41, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Oliver, R.J.; Macdonald, D.G.; Felix, D.H. Aspects of cell proliferation in oral epithelial dysplastic lesions. J. Oral Pathol. Med. 2000, 29, 49–55. [Google Scholar] [CrossRef]
- Alvarado, C.G.; Maruyama, S.; Cheng, J.; Ida-Yonemochi, H.; Kobayashi, T.; Yamazaki, M.; Takagi, R.; Saku, T. Nuclear translocation of beta-catenin synchronized with loss of E-cadherin in oral epithelial dysplasia with a characteristic two-phase appearance. Histopathology 2011, 59, 283–291. [Google Scholar] [CrossRef]
- Lo Muzio, L.; Lo Russo, L.; Falaschini, S.; Ciavarella, D.; Pentenero, M.; Arduino, P.; Favia, G.; Maiorano, E.; Rubini, C.; Pieramici, T.; et al. beta- and gamma-catenin expression in oral dysplasia. Oral. Oncol. 2009, 45, 501–504. [Google Scholar] [CrossRef]
- Elferink, L.A.; Resto, V.A. Receptor-tyrosine-kinase-targeted therapies for head and neck cancer. J. Signal Transduct. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.-Y.; Tsai, S.-Y.; Chen, S.-H.; Tsou, H.; Yen, C.-J.; Liu, K.-J.; Fang, H.-L.; Wu, H.-C.; Chuang, B.-F.; Chou, S.-W.; et al. Dissecting the EGFR-PI3K-AKT pathway in oral cancer highlights the role of the EGFR variant III and its clinical relevance. J. Biomed. Sci. 2013, 20, 43. [Google Scholar] [CrossRef] [Green Version]
- Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [Google Scholar] [CrossRef]
- Sophia, J.; Kowshik, J.; Mishra, R.; Nagini, S. Nimbolide, a neem limonoid inhibits phosphatidyl inositol-3 kinase to activate glycogen synthase kinase-3β in a hamster model of oral oncogenesis. Sci. Rep. 2016, 6, 22192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Li, C. Convergence between Wnt-beta-catenin and EGFR signaling in cancer. Mol. Cancer 2010, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Hung, H.-W.; Hung, P.-H.; Shieh, Y.-S. Epidermal growth factor receptor regulates β-catenin location, stability, and transcriptional activity in oral cancer. Mol. Cancer 2010, 9, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaw, S.; Majeed, A.A.; Dalley, A.; Chan, A.; Stein, S.; Farah, C.S. Epithelial to mesenchymal transition (EMT) biomarkers–E-cadherin, beta-catenin, APC and Vimentin–in oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Zaid, K.W. Immunohistochemical assessment of E-cadherin and beta-catenin in the histological differentiations of oral squamous cell carcinoma. Asian Pac. J. Cancer Prev. 2014, 15, 8847–8853. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Xu, Q.; Zhang, X.; Van Brunt, L.A.; Ticha, P.; Helms, J. Wnt-responsive stem cell fates in the oral mucosa. iScience 2019, 21, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Fisher, A.V.; Yin, Y.; Maruyama, T.; Veith, G.M.; Dhandha, M.; Huang, G.J.; Hsu, W.; Ma, L. The inductive role of Wnt-β-Catenin signaling in the formation of oral apparatus. Dev. Boil. 2011, 356, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Pannone, G.; Bufo, P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G.; Serpico, R.; Cafarelli, B.; Abbruzzese, A.; et al. WNT pathway in oral cancer: Epigenetic inactivation of WNT-inhibitors. Oncol. Rep. 2010, 24, 1035–1041. [Google Scholar]
- Lyakhovitsky, A.; Barzilai, A.; Fogel, M.; Trau, H.; Huszar, M. Expression of e-cadherin and beta-catenin in cutaneous squamous cell carcinoma and its precursors. Am. J. Dermatopathol. 2004, 26, 372–378. [Google Scholar] [CrossRef]
- Shiah, S.-G.; Hsiao, J.-R.; Chang, W.-M.; Chen, Y.-W.; Jin, Y.-T.; Wong, T.-Y.; Huang, J.-S.; Tsai, S.-T.; Hsu, Y.-M.; Chou, S.-T.; et al. Downregulated miR329 and miR410 promote the proliferation and invasion of oral squamous cell carcinoma by targeting Wnt-7b. Cancer Res. 2014, 74, 7560–7572. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Ma, Y.; Li, J.; Chen, H.; Xie, Y.; Chen, M.; Zhao, X.; Tang, S.; Zhao, S.; Zhang, Y.; et al. WNT7A promotes EGF-induced migration of oral squamous cell carcinoma cells by activating β-catenin/MMP9-mediated signaling. Front. Pharmacol. 2020, 11, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritta’, M.; De Andrea, M.; Mondini, M.; Mazibrada, J.; Giordano, C.; Pecorari, G.; Garzaro, M.; Landolfo, V.; Schena, M.; Chiusa, L.; et al. Cell cycle and viral and immunologic profiles of head and neck squamous cell carcinoma as predictable variables of tumor progression. Head Neck 2009, 31, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Basnaker, M.; Sp, S.; Bnvs, S. Cyclin d1 gene expression in oral mucosa of tobacco chewers"-an immunohistochemical study. J. Clin. Diagn. Res. 2014, 8, ZC70. [Google Scholar] [CrossRef]
- Perisanidis, B.; Wrba, F.; Brandstetter, A.; El Gazzar, S.; Papadogeorgakis, N.; Kyzas, P.A.; Filipits, M.; Perisanidis, C.; Seemann, R.; Ewers, R. Evaluation of immunohistochemical expression of p53, p21, p27, cyclin D1, and Ki67 in oral and oropharyngeal squamous cell carcinoma. J. Oral Pathol. Med. 2012, 41, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Olimid, D.A.; Simionescu, C.E.; Mărgăritescu, C.; Florescu, A. Immunoexpression of Ki67 and cyclin D1 in oral squamous carcinomas. Rom. J. Morphol. Embryol. 2012, 53 (Suppl. 3), 795–798. [Google Scholar] [PubMed]
- Negi, A.; Puri, A.; Gupta, R.; Nangia, R.; Sachdeva, A.; Mittal, M. Comparison of immunohistochemical expression of antiapoptotic protein survivin in normal oral mucosa, oral leukoplakia, and oral squamous cell carcinoma. Pathol. Res. Int. 2015, 2015, 1–6. [Google Scholar] [CrossRef]
- Deo, P.N.; Deshmukh, R. Expression of survivin in dysplasia and different grades of oral squamous cell carcinoma. Transl. Res. Oral Oncol. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, Y.; Ding, Y.; Tang, X.; Sun, Z. Impacts of survivin and caspase-3 on apoptosis and angiogenesis in oral cancer. Oncol. Lett. 2017, 14, 3774–3779. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, M.; Jewell, S. Quantitative estimation of PCNA, c-myc, EGFR and TGF-α in oral submucous fibrosis—An immunohistochemical study. Oral Oncol. 2001, 37, 461–467. [Google Scholar] [CrossRef]
- Pallavi, N.; Nalabolu, G.R.K.; Hiremath, S.K.S. Bcl-2 and c-Myc expression in oral dysplasia and oral squamous cell carcinoma: An immunohistochemical study to assess tumor progression. J. Oral Maxillofac. Pathol. 2018, 22, 325–331. [Google Scholar] [CrossRef]
- Voronkov, A.; Krauss, S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr. Pharm. Des. 2013, 19, 634–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, H.; Sasaki, S.; Suzuki, H.; Toyota, M.; Maruyama, R.; Nojima, M.; Yamamoto, H.; Omata, M.; Tokino, T.; Imai, K.; et al. Frequent epigenetic inactivation of SFRP genes in hepatocellular carcinoma. J. Gastroenterol. 2008, 43, 378–389. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, Y.S.; Chung, J.H. Epigenetic silencing of the WNT antagonist DICKKOPF-1 in cervical cancer cell lines. Gynecol. Oncol. 2008, 109, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-W.; Chung, M.-T.; Lai, H.-C.; De Yan, M.; Shih, Y.-L.; Chang, C.-C.; Yu, M.-H. Methylation analysis of SFRP genes family in cervical adenocarcinoma. J. Cancer Res. Clin. Oncol. 2009, 135, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Rawson, J.B.; Manno, M.; Mrkonjic, M.; Daftary, D.; Dicks, E.; Buchanan, D.; Younghusband, H.B.; Parfrey, P.S.; Young, J.; Pollett, A.; et al. Promoter methylation of Wnt antagonists DKK1 and SFRP1 is associated with opposing tumor subtypes in two large populations of colorectal cancer patients. Carcinogenesis 2011, 32, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.; Wang, L.; Zhu, L.; Zhang, C.; Zhou, J. Secreted frizzled-related protein 2 is epigenetically silenced and functions as a tumor suppressor in oral squamous cell carcinoma. Mol. Med. Rep. 2014, 10, 2293–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluszczak, J.; Sarbak, J.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Jarmuz-Szymczak, M.; Grenman, R.; Mielcarek-Kuchta, D.; Baer-Dubowska, W. The negative regulators of Wnt pathway-DACH1, DKK1, and WIF1 are methylated in oral and oropharyngeal cancer and WIF1 methylation predicts shorter survival. Tumor Boil. 2015, 36, 2855–2861. [Google Scholar] [CrossRef] [Green Version]
- Miaczynska, M.; Pelkmans, L.; Zerial, M. Not just a sink: Endosomes in control of signal transduction. Curr. Opin. Cell Boil. 2004, 16, 400–406. [Google Scholar] [CrossRef]
- Dobrowolski, R.; De Robertis, E.M. Endocytic control of growth factor signalling: Multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Boil. 2012, 13, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Blitzer, J.T.; Nusse, R. A critical role for endocytosis in Wnt signaling. BMC Cell Boil. 2006, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, A.I.; Kurz, J.; Kauffeld, S.; Chen, Q.; Reeves, P.M.; Weber, S.; Schindler, S.; Davidson, G.; Kirchhausen, T.; Scholpp, S. In vivo analysis of formation and endocytosis of the Wnt/beta-catenin signaling complex in zebrafish embryos. J. Cell Sci. 2014, 127, 3970–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, A.; Yamamoto, H. Regulation of Wnt signalling by receptor-mediated endocytosis. J. Biochem. 2007, 141, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Komekado, H.; Kikuchi, A. caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of β-catenin. Dev. Cell 2006, 11, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.-L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosesson, Y.; Mills, G.B.; Yarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850. [Google Scholar] [CrossRef]
- Lanzetti, L.; Di Fiore, P.P. Behind the scenes: Endo/exocytosis in the acquisition of metastatic traits. Cancer Res. 2017, 77, 1813–1817. [Google Scholar] [CrossRef] [Green Version]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Boil. 2013, 5, a016949. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, H.A. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Boil. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Binns, D.; Januszewski, T.; Chen, Y.; Hill, J.; Markin, V.S.; Zhao, Y.; Gilpin, C.; Chapman, K.D.; Anderson, R.G.; Goodman, J.M. An intimate collaboration between peroxisomes and lipid bodies. J. Cell Boil. 2006, 173, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, N.D.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic–the update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.C.; Caswell, P.T.; Norman, J.C. Endocytic recycling pathways: Emerging regulators of cell migration. Curr. Opin. Cell Boil. 2006, 18, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nature 2015, 17, 1412–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- . Palamidessi, A.; Frittoli, E.; Ducano, N.; Offenhauser, N.; Sigismund, S.; Kajiho, H.; Parazzoli, D.; Oldani, A.; Gobbi, M.; Serini, G.G.; et al. The GTPase-activating protein RN-TRE controls focal adhesion turnover and cell migration. Curr. Boil. 2013, 23, 2355–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza, P.; Ortiz, R.; Díaz, J.; Quest, A.F.G.; Leyton, L.; Stupack, D.; Torres, V.A. Rab5 activation promotes focal adhesion disassembly, migration and invasiveness in tumor cells. J. Cell Sci. 2013, 126, 3835–3847. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, P.A.; Silva, P.; Díaz, J.; Arriagada, C.; Canales, J.; Cerda, O.; Torres, V.A. Calpain2 mediates Rab5-driven focal adhesion disassembly and cell migration. Cell Adhes. Migr. 2018, 12, 185–194. [Google Scholar] [CrossRef]
- Palamidessi, A.; Frittoli, E.; Garrè, M.; Faretta, M.; Mione, M.C.; Testa, I.; Diaspro, A.; Lanzetti, L.; Scita, G.; Di Fiore, P.P. Endocytic trafficking of rac is required for the spatial restriction of signaling in cell migration. Cell 2008, 134, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Díaz, J.; Mendoza, P.; Ortiz, R.; Díaz, N.; Leyton, L.; Stupack, D.; Quest, A.F.; Torres, V.A. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J. Cell Sci. 2014, 127, 2401–2406. [Google Scholar] [CrossRef] [Green Version]
- Frittoli, E.; Palamidessi, A.; Marighetti, P.; Confalonieri, S.; Bianchi, F.; Malinverno, C.; Mazzarol, G.; Viale, G.; Martin-Padura, I.; Garré, M.; et al. A RAB5/RAB4 recycling circuitry induces a proteolytic invasive program and promotes tumor dissemination. J. Cell Biol. 2014, 206, 307–328. [Google Scholar] [CrossRef] [Green Version]
- Serio, G.; Margaria, V.; Jensen, S.; Oldani, A.; Bartek, J.; Bussolino, F.; Lanzetti, L. Small GTPase Rab5 participates in chromosome congression and regulates localization of the centromere-associated protein CENP-F to kinetochores. Proc. Natl. Acad. Sci. USA 2011, 108, 17337–17342. [Google Scholar] [CrossRef] [Green Version]
- Dey, K.K.; Pal, I.; Bharti, R.; Dey, G.; Kumar, B.N.P.; Rajput, S.; Parekh, A.; Parida, S.; Halder, P.; Kulavi, I.; et al. Identification of RAB2A and PRDX1 as the potential biomarkers for oral squamous cell carcinoma using mass spectrometry-based comparative proteomic approach. Tumor Boil. 2015, 36, 9829–9837. [Google Scholar] [CrossRef]
- Da Silva, S.D.; Marchi, F.A.; Xu, B.; Bijian, K.; Alobaid, F.; Mlynarek, A.; Rogatto, S.R.; Hier, M.; Kowalski, L.P.; Alaoui-Jamali, M.A. Predominant Rab-GTPase amplicons contributing to oral squamous cell carcinoma progression to metastasis. Oncotarget 2015, 6, 21950–21963. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Miyazawa, H.; Kobayashi, H.; Noguchi, N.; Lambert, D.; Kawashiri, S. Caveolin-1 expression at metastatic lymph nodes predicts unfavorable outcome in patients with oral squamous cell carcinoma. Pathol. Oncol. Res. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-J.; Chang, K.-P.; Chang, Y.-J.; Hsu, C.-W.; Liang, Y.; Yu, J.-S.; Chi, L.-M.; Chang, Y.-S.; Wu, C.-C. Identification of guanylate-binding protein 1 as a potential oral cancer marker involved in cell invasion using OMICS-based analysis. J. Proteome Res. 2011, 10, 3778–3788. [Google Scholar] [CrossRef]
- Amornphimoltham, P.; Rechache, K.; Thompson, J.; Masedunskas, A.; Leelahavanichkul, K.; Patel, V.; Molinolo, A.; Gutkind, J.S.; Weigert, R. Rab25 regulates invasion and metastasis in head and neck cancer. Clin. Cancer Res. 2013, 19, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, M.J.A.M.; Melchers, L.J.; Mastik, M.F.; Slagter-Menkema, L.; Groen, H.J.M.; Van Der Laan, B.F.A.M.; Van Criekinge, W.; De Meyer, T.; Denil, S.; Van Der Vegt, B.; et al. RAB25 expression is epigenetically downregulated in oral and oropharyngeal squamous cell carcinoma with lymph node metastasis. Epigenetics 2016, 11, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Lu, C.; Ai, H. Rab5a is overexpressed in oral cancer and promotes invasion through ERK/MMP signaling. Mol. Med. Rep. 2017, 16, 4569–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.Y.; Keller, T.H. The use of porcupine inhibitors to target Wnt-driven cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5472–5476. [Google Scholar] [CrossRef]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2015, 35, 2197–2207. [Google Scholar] [CrossRef] [Green Version]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Madan, B.; Virshup, D.M. Targeting Wnts at the source-new mechanisms, new biomarkers, new drugs. Mol. Cancer Ther. 2015, 14, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Heuvel, M.V.D.; Harryman-Samos, C.; Klingensmith, J.; Perrimon, N.; Nusse, R. Mutations in the segment polarity genes wingless and porcupine impair secretion of the wingless protein. EMBO J. 1993, 12, 5293–5302. [Google Scholar] [CrossRef] [PubMed]
- Herr, P.; Basler, K. Porcupine-mediated lipidation is required for Wnt recognition by Wls. Dev. Boil. 2012, 361, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, M.; Flores, T.; Betancur, D.; Peña-Oyarzún, D.; Torres, V.A. Wnt/β-Catenin Signaling in Oral Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 4682. https://doi.org/10.3390/ijms21134682
Reyes M, Flores T, Betancur D, Peña-Oyarzún D, Torres VA. Wnt/β-Catenin Signaling in Oral Carcinogenesis. International Journal of Molecular Sciences. 2020; 21(13):4682. https://doi.org/10.3390/ijms21134682
Chicago/Turabian StyleReyes, Montserrat, Tania Flores, Diego Betancur, Daniel Peña-Oyarzún, and Vicente A. Torres. 2020. "Wnt/β-Catenin Signaling in Oral Carcinogenesis" International Journal of Molecular Sciences 21, no. 13: 4682. https://doi.org/10.3390/ijms21134682
APA StyleReyes, M., Flores, T., Betancur, D., Peña-Oyarzún, D., & Torres, V. A. (2020). Wnt/β-Catenin Signaling in Oral Carcinogenesis. International Journal of Molecular Sciences, 21(13), 4682. https://doi.org/10.3390/ijms21134682