Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes



Abstract
:1. Introduction
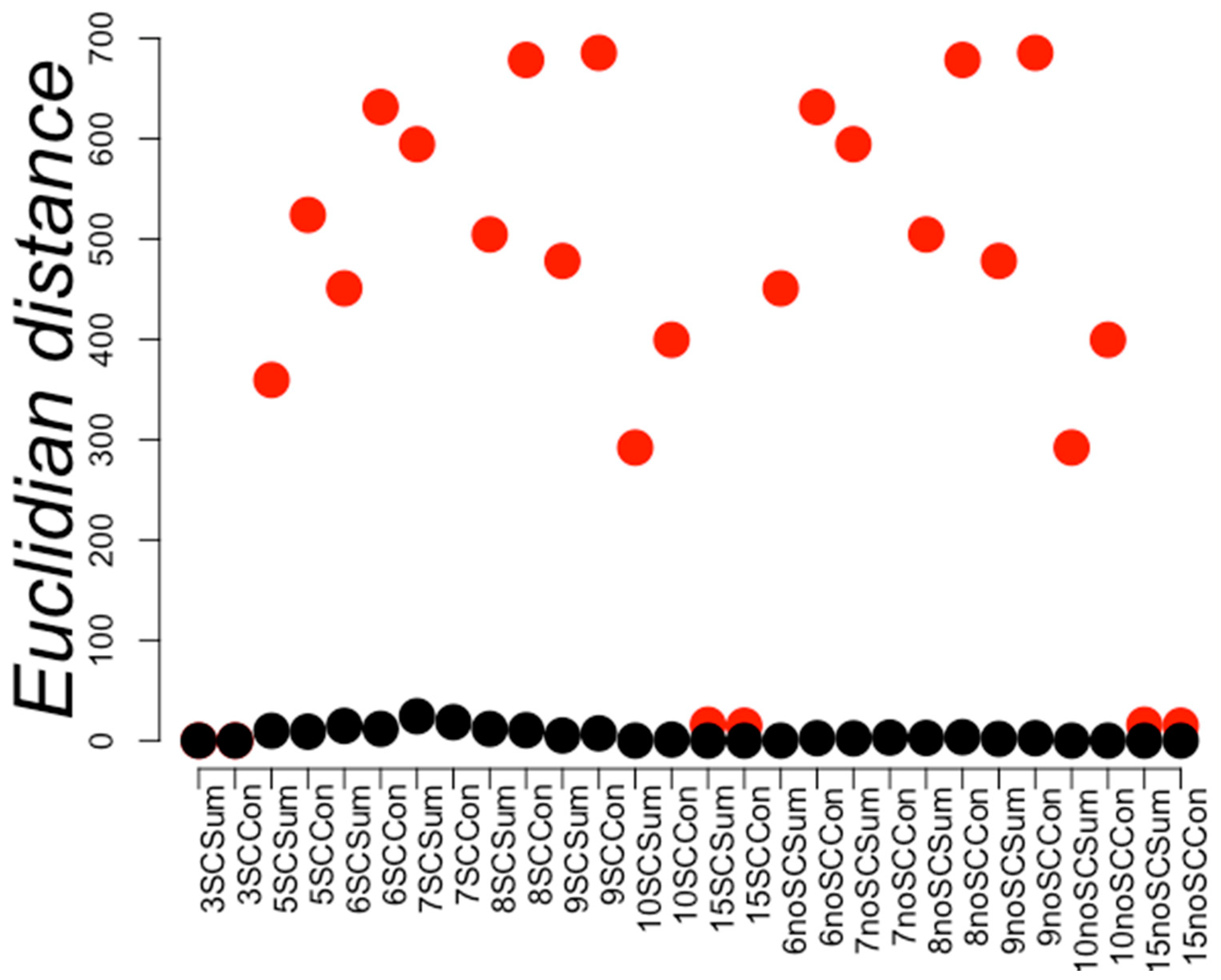
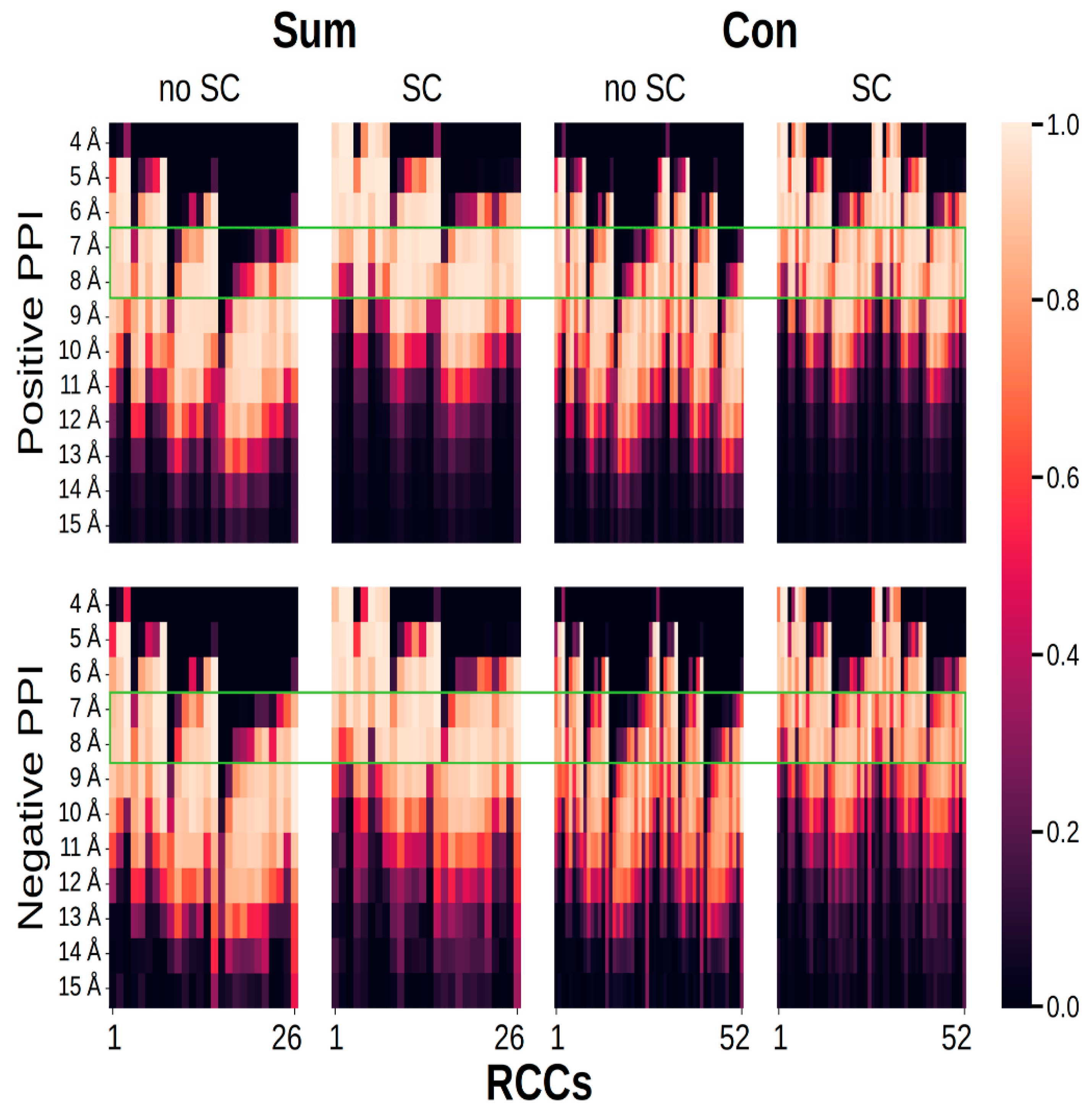
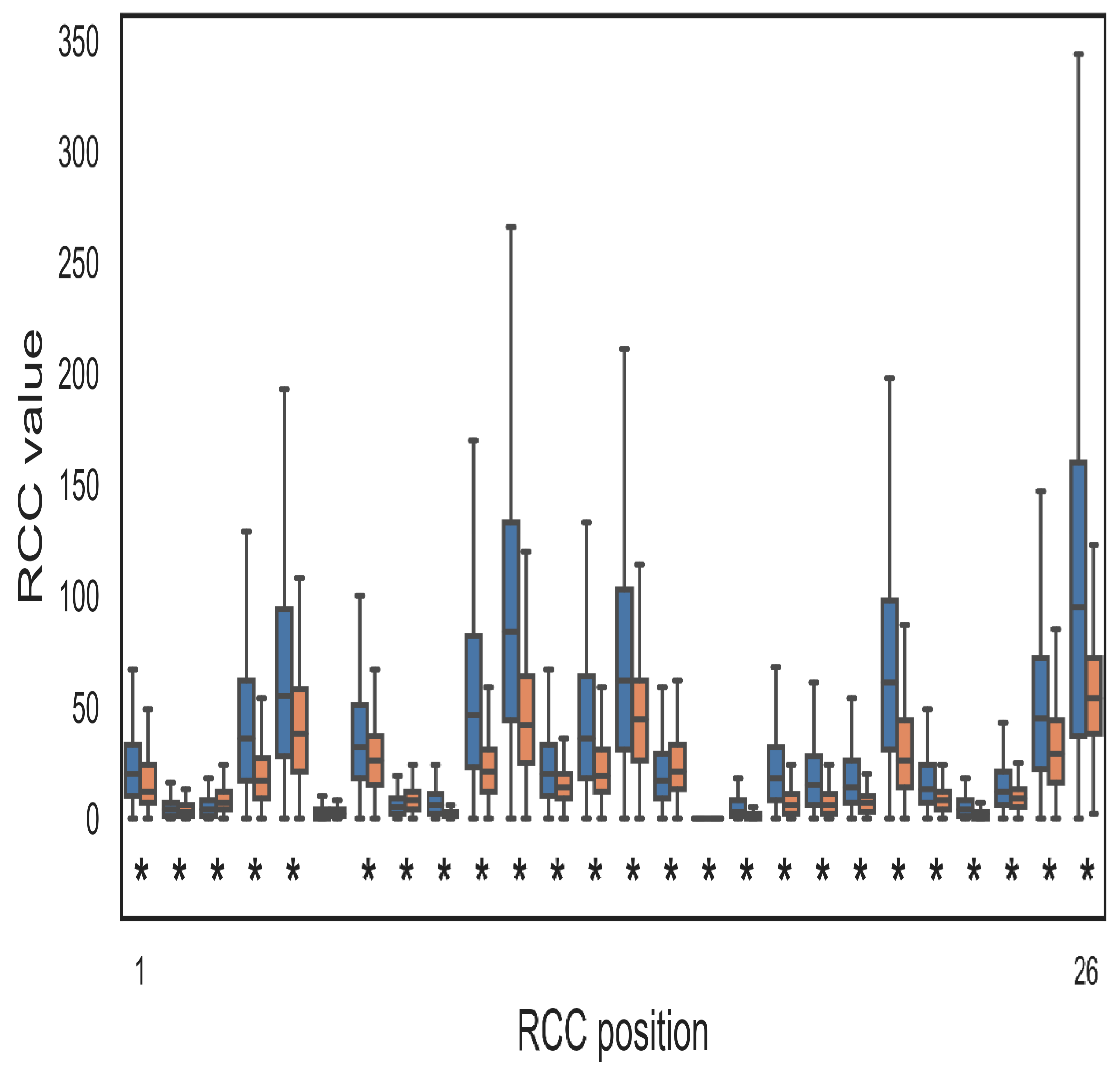
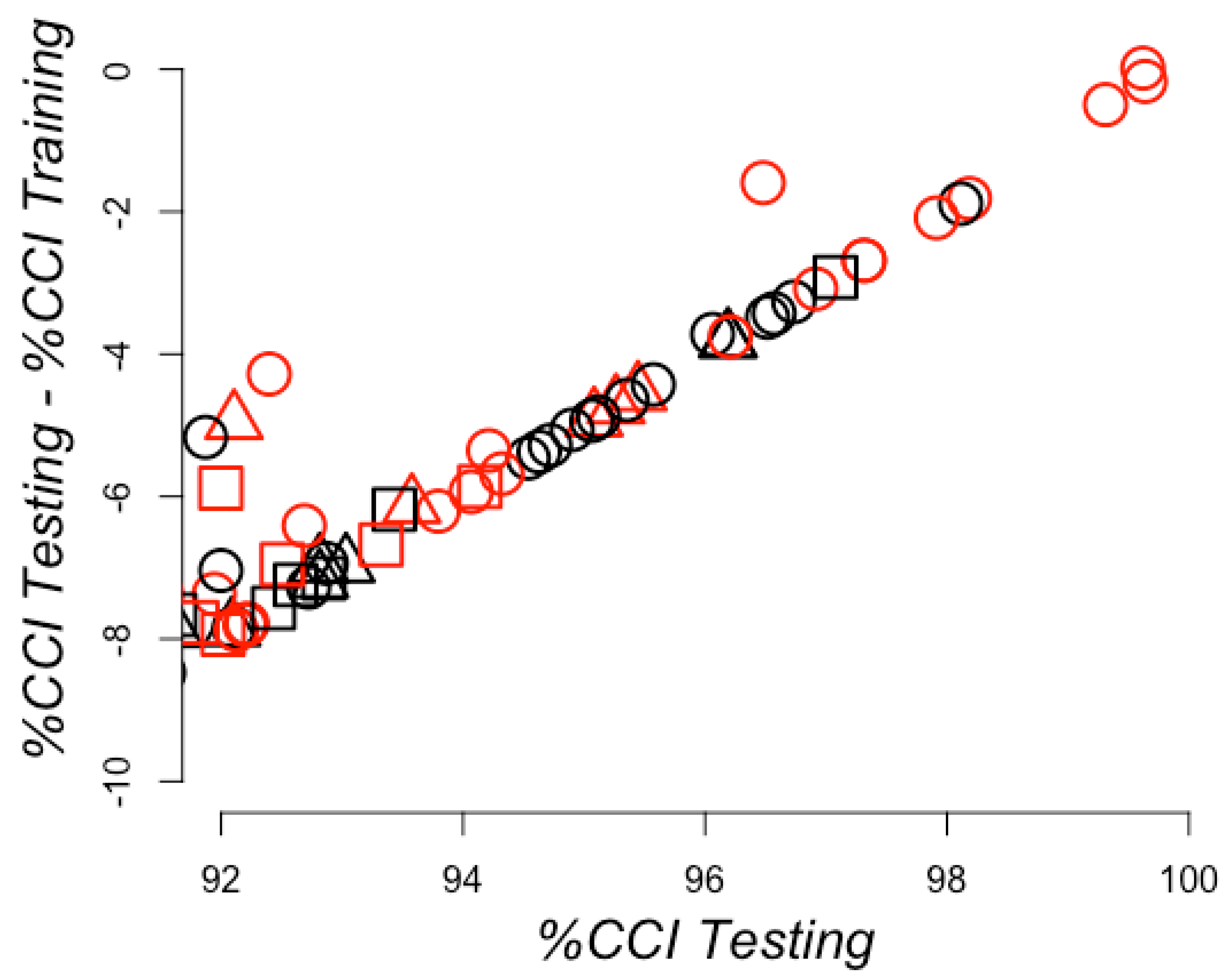
2. Results
3. Discussion
4. Materials and Methods
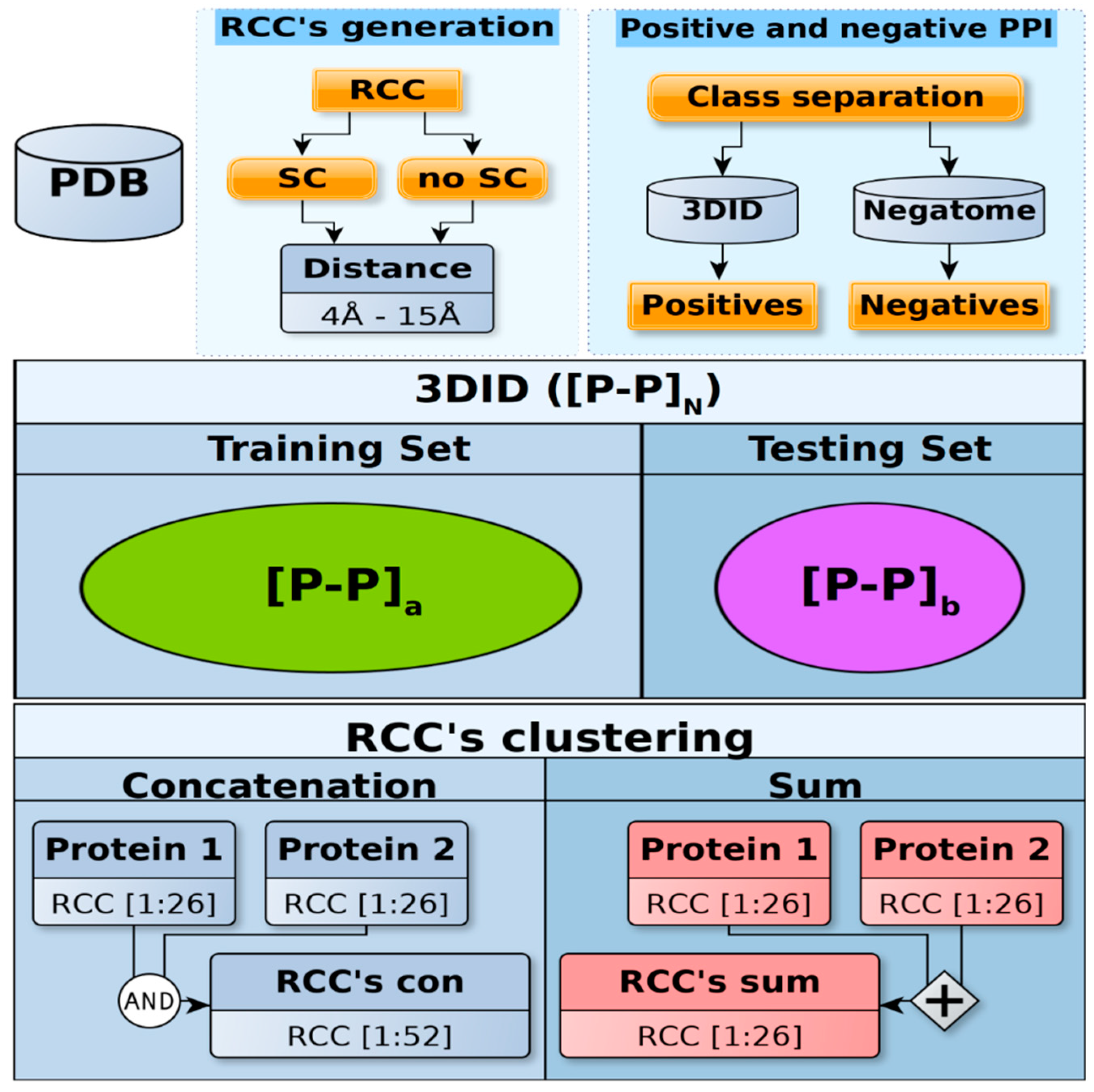
4.1. Datasets
4.2. Machine Learning and Statistical Testing
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.P.; Liu, B.; Sun, Y.; Chiang, V.L.; Sederoff, R.R. Enzyme-enzyme interactions in monolignol biosynthesis. Front Plant Sci. 2019, 9, 1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freilich, R.; Arhar, T.; Abrams, J.L.; Gestwicki, J.E. Protein-Protein Interactions in the Molecular Chaperone Network. Acc. Chem. Res. 2018, 51, 940–949. [Google Scholar] [CrossRef]
- Zahiri, J.; Emamjomeh, A.; Bagheri, S.; Ivazeh, A.; Mahdevar, G.; Sepasi Tehrani, H.; Mirzaie, M.; Fakheri, B.A.; Mohammad-Noori, M. Protein complex prediction: A survey. Genomics 2020, 112, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, C.; Deng, L. Machine learning approaches for protein-protein interaction hot spot prediction: Progress and comparative assessment. Molecules 2018, 23, 2535. [Google Scholar] [CrossRef] [Green Version]
- Kotlyar, M.; Rossos, A.E.M.; Jurisica, I. Prediction of Protein-Protein Interactions. Curr. Protoc. Bioinform. 2017, 60, 8.2.1–8.2.14. [Google Scholar] [CrossRef]
- Bzdok, D.; Krzywinski, M.; Altman, N. Points of significance: Machine learning: Supervised methods. Nat. Methods 2018, 15, 5–6. [Google Scholar] [CrossRef]
- Ruiz-Blanco, Y.B.; Paz, W.; Green, J.; Marrero-Ponce, Y. ProtDCal: A program to compute general-purpose-numerical descriptors for sequences and 3D-structures of proteins. BMC Bioinform. 2015, 16, 162. Available online: https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-015-0586-0 (accessed on 3 February 2020). [CrossRef] [Green Version]
- Shen, H.B.; Chou, K.C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef]
- Li, Z.R.; Lin, H.H.; Han, L.Y.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, w32–w37. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, D.; Saha, S. Machine-learning techniques for the prediction of protein–protein interactions. J. Biosci. 2019, 44, 104. [Google Scholar] [CrossRef]
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Green, J.R.; Sanchez-Garcia, E. ProtDCal-Suite: A web server for the numerical codification and functional analysis of proteins. Protein Sci. 2019, 28, 1734–1743. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31271472 (accessed on 3 February 2020). [CrossRef] [PubMed]
- Chen, M.; Ju, C.J.T.; Zhou, G.; Chen, X.; Zhang, T.; Chang, K.W.; Zaniolo, C.W.; Wang, W. Multifaceted Protein-Protein Interaction Prediction Based on Siamese Residual RCNN. Bioinformatics 2019, 35, i305–i314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Chan, K.C.C. Extracting Coevolutionary Features from Protein Sequences for Predicting Protein-Protein Interactions. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Ardakani, F.B.; Schmidt, F.; Schulz, M.H. Predicting transcription factor binding using ensemble random forest models [version 2; peer review: 2 approved]. F1000Research 2019, 7, 1603. [Google Scholar] [CrossRef]
- Hue, M.; Riffle, M.; Vert, J.P.; Noble, W.S. Large-scale prediction of protein-protein interactions from structures. BMC Bioinform. 2010, 11, 144. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.W.; Zhou, Y.Q.; Ul Qamar, M.T.; Chen, L.L.; Ding, Y.D. Prediction of protein–protein interactions by evidence combining methods. Int. J. Mol. Sci. 2016, 17, 1946. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Kihara, D. Computational Methods for Predicting Protein-Protein Interactions Using Various Protein Features. Curr. Protoc. Protein Sci. 2018, 93, e62. Available online: http://doi.wiley.com/10.1002/cpps.62 (accessed on 3 February 2020). [CrossRef]
- Zhang, S.B.; Tang, Q.R. Protein-protein interaction inference based on semantic similarity of Gene Ontology terms. J. Theor. Biol. 2016, 401, 30–37. [Google Scholar] [CrossRef]
- Corral-Corral, R.; Chavez, E.; Del Rio, G. Machine Learnable Fold Space Representation based on Residue Cluster Classes. Comput. Biol. Chem. 2015, 59, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosca, R.; Céol, A.; Stein, A.; Olivella, R.; Aloy, P. 3did: A catalog of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2014, 42, D374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blohm, P.; Frishman, G.; Smialowski, P.; Goebels, F.; Wachinger, B.; Ruepp, A.; Frishman, D. Negatome 2.0: A database of non-interacting proteins derived by literature mining, manual annotation and protein structure analysis. Nucleic Acids Res. 2014, 42, D396–D400. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24214996 (accessed on 16 February 2020). [CrossRef] [PubMed]
- Atkeson, C.G.; Moore, A.W.; Schaal, S. Locally Weighted Learning. Artif. Intell. Rev. 1997, 11, 11–73. [Google Scholar] [CrossRef]
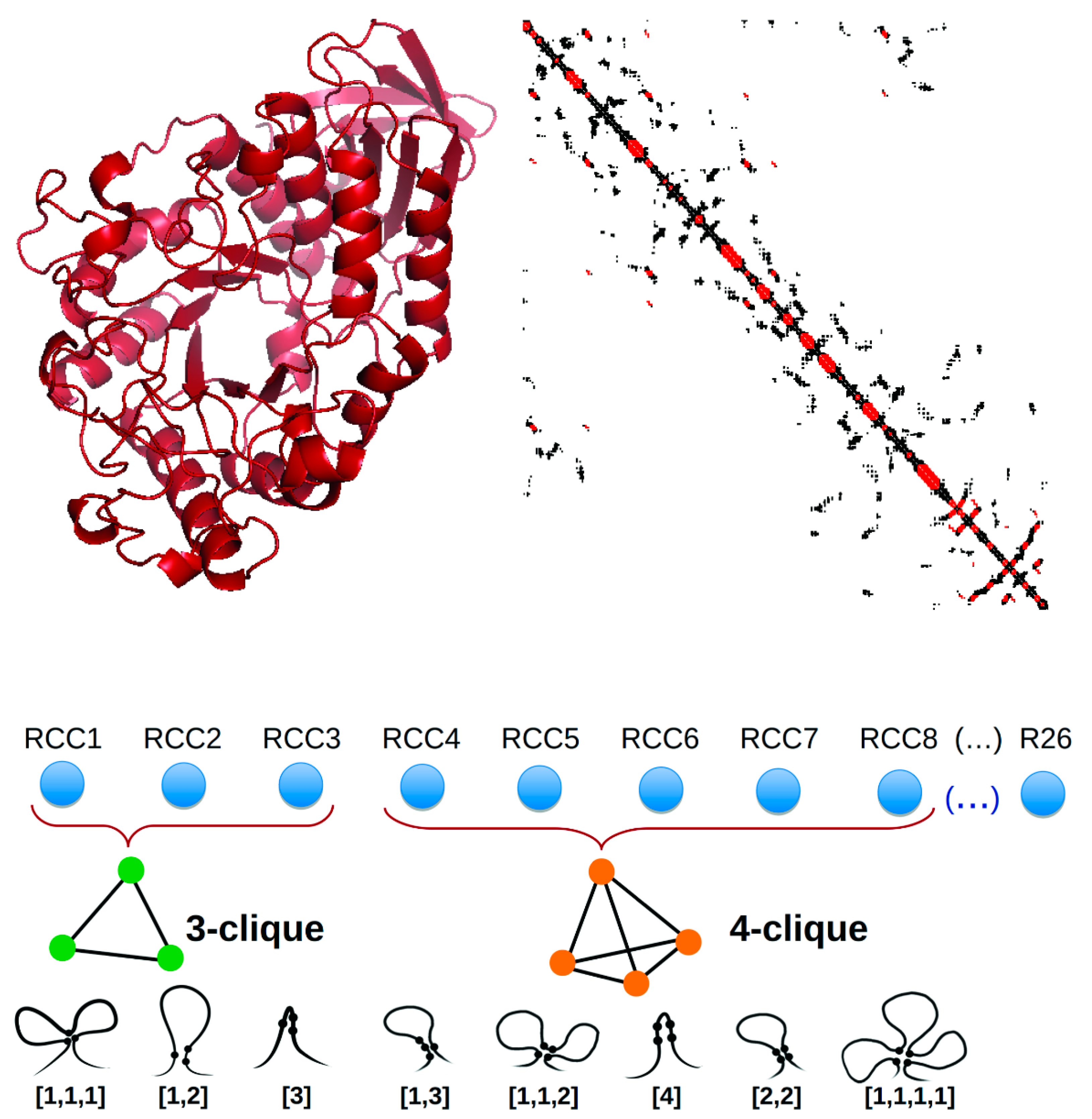
- Fontove, F.; Del Rio, G. Residue cluster classes: A unified protein representation for efficient structural and functional classification. Entropy 2020, 22, 472. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef]
- Elefsinioti, A.; Saraç, Ö.S.; Hegele, A.; Plake, C.; Hubner, N.C.; Poser, I.; Sarov, M.; Hyman, A.; Mann, M.; Schroeder, M.; et al. Large-scale de novo prediction of physical protein-protein association. Mol. Cell. Proteomics 2011, 10, M111.010629. [Google Scholar] [CrossRef] [Green Version]
- Petschnigg, J.; Groisman, B.; Kotlyar, M.; Taipale, M.; Zheng, Y.; Kurat, C.F.; Sayad, A.; Sierra, J.R.; Usaj, M.M.; Snider, J.; et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 2014, 11, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.S.; Yu, J.; Gardenour, K.R.; Finley, R.L.; Ideker, T. Cost-effective strategies for completing the interactome. Nat. Methods 2009, 6, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Ben-Hur, A.; Noble, W.S. Choosing negative examples for the prediction of protein-protein interactions. BMC Bioinform. 2006, 7, S2. [Google Scholar] [CrossRef] [Green Version]
- Hamp, T.; Rost, B. More challenges for machine-learning protein interactions. Bioinformatics 2015, 31, 1521–1525. Available online: https://academic.oup.com/bioinformatics/article-lookup/doi/10.1093/bioinformatics/btu857 (accessed on 19 February 2020). [CrossRef] [PubMed] [Green Version]
- Park, Y.; Marcotte, E.M. Flaws in evaluation schemes for pair-input computational predictions. Nat. Methods 2012, 9, 1134–1136. [Google Scholar] [CrossRef] [PubMed]
- Basile, W.; Sachenkova, O.; Light, S.; Elofsson, A. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput. Biol. 2017, 13, e1005375. Available online: https://dx.plos.org/10.1371/journal.pcbi.1005375 (accessed on 1 April 2020). [CrossRef] [PubMed]
- Kotlyar, M.; Pastrello, C.; Sheahan, N.; Jurisica, I. Integrated interactions database: Tissue-specific view of the human and model organism interactomes. Nucleic Acids Res. 2016, 44, D536–D541. Available online: https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/gkv1115 (accessed on 19 February 2020). [CrossRef] [PubMed]
- Snider, J.; Kotlyar, M.; Saraon, P.; Yao, Z.; Jurisica, I.; Stagljar, I. Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 2015, 11, 848. Available online: https://onlinelibrary.wiley.com/doi/abs/10.15252/msb.20156351 (accessed on 19 February 2020). [CrossRef] [PubMed]
- Wang, Z.; Clark, N.R.; Ma’ayan, A. Dynamics of the discovery process of protein-protein interactions from low content studies. BMC Syst. Biol. 2015, 9, 26. Available online: https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-015-0173-z (accessed on 19 February 2020). [CrossRef] [PubMed] [Green Version]
- Fischer, H.; Polikarpov, I.; Craievich, A.F. Average protein density is a molecular-weight-dependent function. Protein Sci. 2009, 13, 2825–2828. [Google Scholar] [CrossRef]
- Kaddis, C.S.; Lomeli, S.H.; Yin, S.; Berhane, B.; Apostol, M.I.; Kickhoefer, V.A.; Rome, L.H.; Loo, J.A. Sizing Large Proteins and Protein Complexes by Electrospray Ionization Mass Spectrometry and Ion Mobility. J. Am. Soc. Mass Spectrom. 2007, 18, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Alquraishi, M.; Valencia, A. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. Available online: http://www.ncbi.nlm.nih.gov/pubmed/31116374 (accessed on 19 February 2020). [CrossRef]
- Roche, D.B.; McGuffin, L.J. Toolbox for protein structure prediction. Methods in Molecular Biology 2016, 1369, 363–377. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. Available online: https://academic.oup.com/nar/article/47/D1/D464/5144139 (accessed on 16 February 2020). [CrossRef] [Green Version]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA Data Mining Software: An Update. ACM SIGKDD Explor. Newsl. 2009, 11, 1. Available online: https://www.kdd.org/exploration_files/p2V11n1.pdf (accessed on 12 February 2020). [CrossRef]
- Kotthoff, L.; Thornton, C.; Hoos, H.H.; Hutter, F.; Leyton-Brown, K. Auto-WEKA 2.0: Automatic model selection and hyperparameter optimization in WEKA. J. Mach. Learn. Res. 2017, 18, 1–5. Available online: http://automl.org/autoweka (accessed on 12 February 2020).
- Mckinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference (SCIPY 2010), Austin, TX, USA, 28 June–3 July 2010; pp. 56–61. [Google Scholar]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; pp. 92–96. Available online: http://statsmodels.sourceforge.net/ (accessed on 5 April 2020).
- Waskom, M. Seaborn: Statistical Data Visualization—Seaborn 0.10.0 Documentation. 2012. Available online: https://seaborn.pydata.org/ (accessed on 9 April 2020).
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 99–104. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P:N | Training | Testing | ||
|---|---|---|---|---|
| P | N | P | N | |
| 1:1 | 692 | 692 | 4819 | 692 |
| 2:1 | 1384 | 692 | 4819 | 692 |
| 3:1 | 2076 | 692 | 4819 | 692 |
| 1:2 | 692 | 1384 | 4819 | 692 |
| 1:3 | 692 | 2076 | 4819 | 692 |
| Training | Testing | ||||
|---|---|---|---|---|---|
| Positives | Negatives | Positives | Negatives | ||
| Concatenation | 1:1 | 489 | 489 | 122 | 122 |
| 2:1 | 978 | 489 | 122 | 122 | |
| 3:1 | 1467 | 489 | 122 | 122 | |
| Sum | 1:1 | 448 | 448 | 111 | 111 |
| 2:1 | 896 | 448 | 111 | 111 | |
| 3:1 | 1344 | 448 | 111 | 111 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poot Velez, A.H.; Fontove, F.; Del Rio, G. Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes. Int. J. Mol. Sci. 2020, 21, 4787. https://doi.org/10.3390/ijms21134787
Poot Velez AH, Fontove F, Del Rio G. Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes. International Journal of Molecular Sciences. 2020; 21(13):4787. https://doi.org/10.3390/ijms21134787
Chicago/Turabian StylePoot Velez, Albros Hermes, Fernando Fontove, and Gabriel Del Rio. 2020. "Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes" International Journal of Molecular Sciences 21, no. 13: 4787. https://doi.org/10.3390/ijms21134787
APA StylePoot Velez, A. H., Fontove, F., & Del Rio, G. (2020). Protein–Protein Interactions Efficiently Modeled by Residue Cluster Classes. International Journal of Molecular Sciences, 21(13), 4787. https://doi.org/10.3390/ijms21134787