Fundamental Mechanisms of Autoantibody-Induced Impairments on Ion Channels and Synapses in Immune-Mediated Cerebellar Ataxias




Abstract
:1. Introduction
- impairment of ion channel(s) or synaptic transmission(s) and the development of clinically evident CAs as a consequence of this (pathogenic action), and
- mimics of these CAs by a passive transfer experiment (passive transfer).
- release machinery proteins
- synaptic adhesion molecules, and
- receptors.
2. Overview of Cerebellar Physiology—Ion Channels and Synaptic Machinery
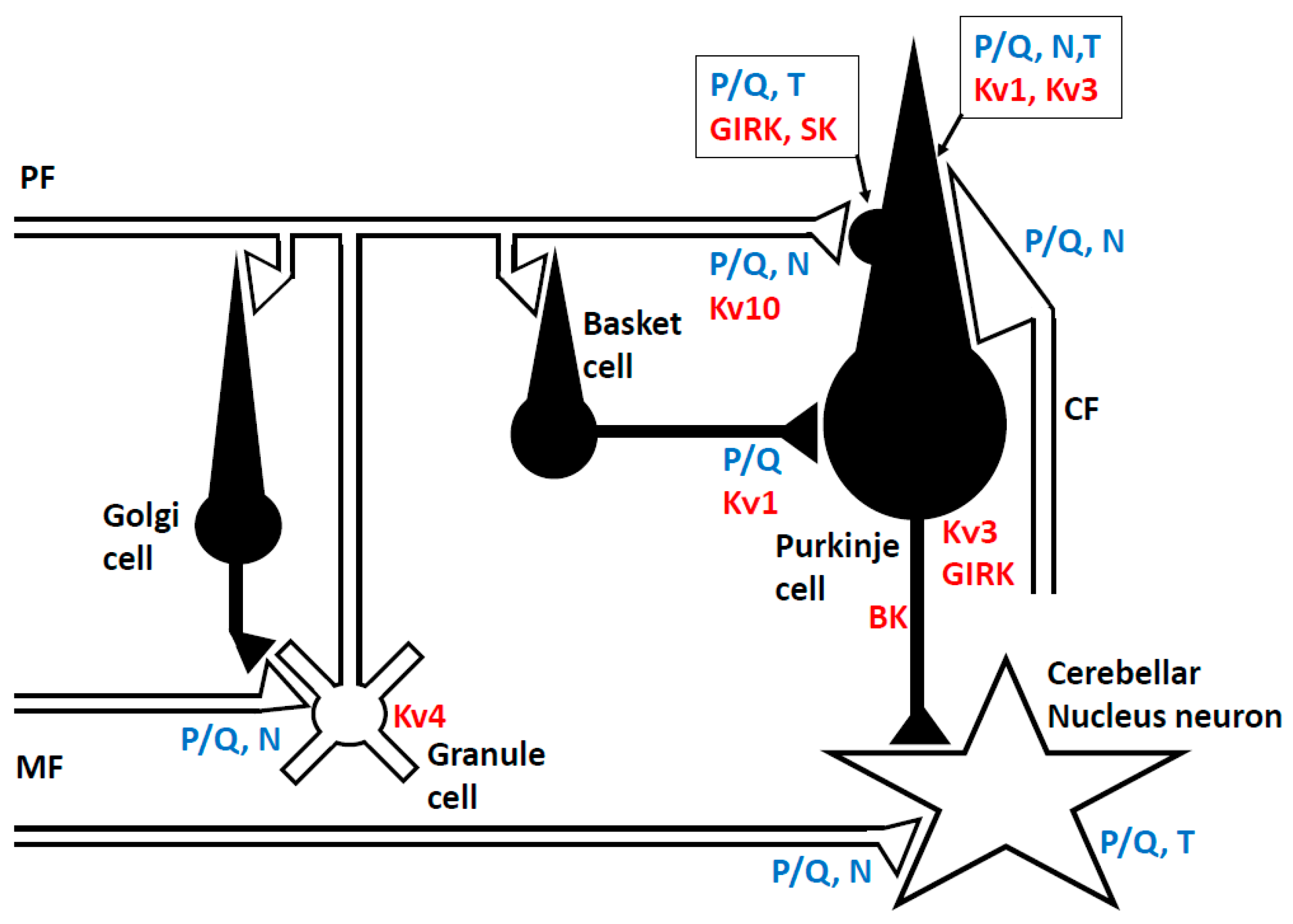
2.1. Neuronal Network in the Cerebellum
2.2. Ion Channels
2.2.1. Sodium (Na+) Channel
2.2.2. Potassium (K+) Channel
2.2.3. Calcium (Ca2+) Channel
2.3. Presynaptic and Inter-Synaptic Machinery
2.3.1. Neurotransmitter Release from the Presynaptic Terminal
2.3.2. Synaptic Adhesion Molecules
2.4. Receptors at the Post-Synaptic Membrane of Purkinje Cells
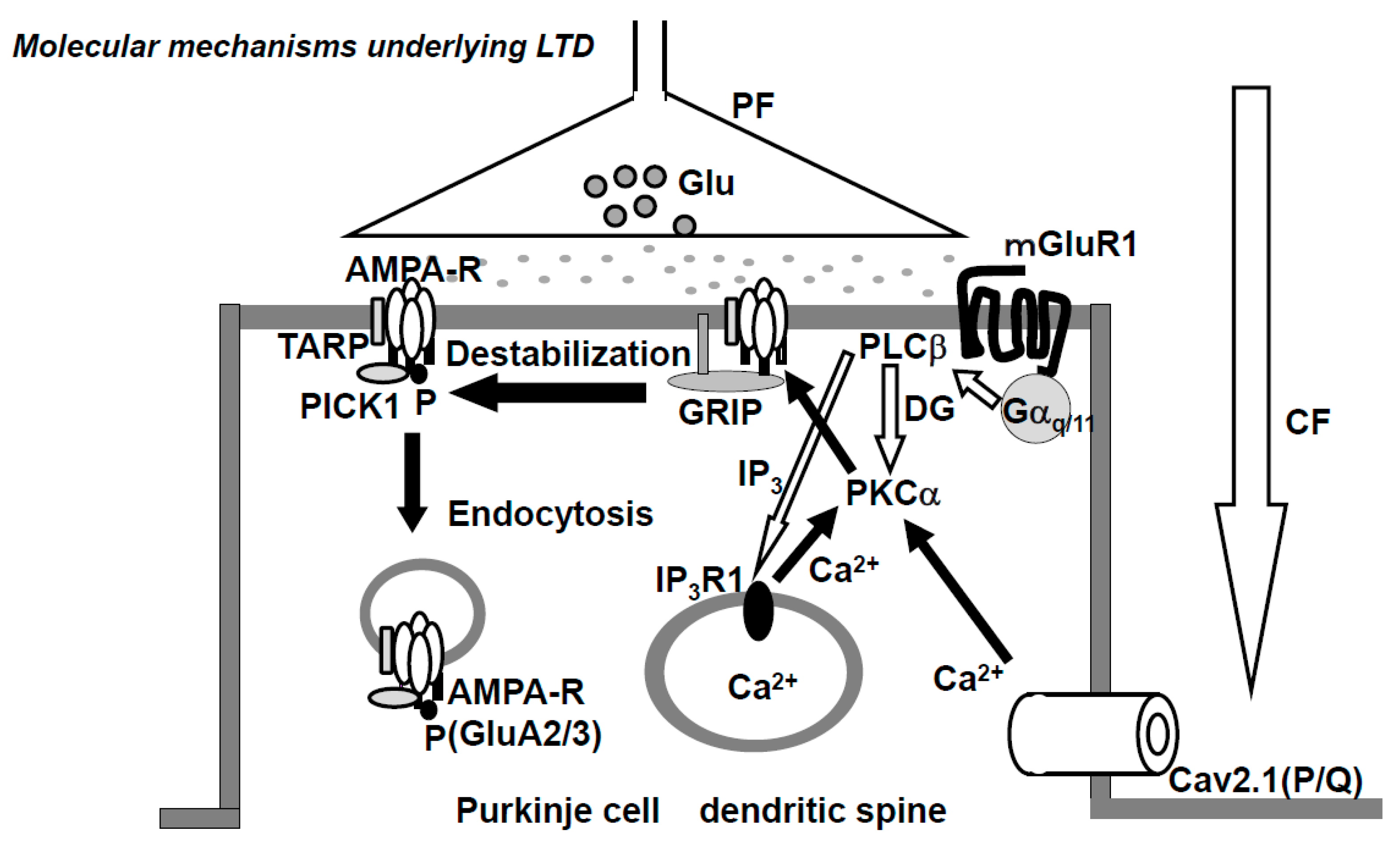
- AMPA (α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid)-type glutamate receptor. The AMPA-type glutamate receptor (AMPAR) is a heteromeric tetramer composed of GluA1-4, where GluA2 and GluA3 are subtypes with short carboxyl (C)-terminus, while GluA1 and GluA4 have long C-terminus [59]. In adults, the only subtypes with short C-terminus (GluA2,3) are expressed in the PF-PC synapse. AMPARs that lack GluA2 or AMPAR composed of GluA2 with non-edited Q/R-site show Ca2+-permeability, but no such Ca2+-permeable AMPARs are expressed in PC [60,61]. The glutamate receptor-interacting protein (GRIP) and transmembrane AMPA receptor regulatory proteins (TARPs), which are expressed at postsynaptic PF-PC synapses, serve as scaffold proteins [62,63] (Figure 2). GRIP, which anchors AMPAR, plays a critical role in neuronal plasticity. Elimination of GRIP1/2 causes the failure of LTD-induction [64]. TARPs modulate the pharmacology and gating of AMPARs.
- NMDA-type glutamate receptor. N-methyl-d aspartate receptors (NMDARs), the ionotropic glutamate receptors endowed with high Ca2+ permeability, play key roles in the induction of LTP and LTD in various brain regions. In adult rodent postsynaptic PCs, NMDARs are expressed at the climbing fiber synapse, but not at the PF-PC synapse [65]. However, NMDARs at the CF-PC synapse are not required for the induction of LTD, rather NMDARs of stellate cells are necessary for LTD induction [66].
- Metabotropic glutamate receptor. The metabotropic glutamate receptor 1 (mGluR1) is expressed in the peripheral zone of the PF-PC synapsis [67]. The cytoskeletal protein, β-III spectrin, is involved in the localization of mGluR1 at the synaptic membrane [68]. Spinocerebellar ataxia 5 (SCA5) is caused by heterozygous mutations in the gene encoding β-III spectrin (SPTBN2). The mGluR1 activates PLCβ and produces IP3 and DG, then IP3 triggers Ca2+-release from the ER. Mutations in mGluR1 are associated with failure of LTD, deficiency of eye-blink conditioning, ataxia, and abnormal synaptic development of the CF-PC synapse [69].
- GABA receptor. Two types of inhibitory interneurons, basket and stellate cells, innervate the PC. Both types of interneurons contain GAD65 for GABA synthesis and packaging of GABA to synaptic vesicles. GABAA receptors (GABAAR) are located on dendritic and somatic membranes of the PC [70]. In PCs, GABAB receptors (GABABR), which are coupled with G proteins, are found on the dendritic membrane in coclusters with GIRK or P/Q-type Ca2+ channel [71]. It should be acknowledged that during the execution of movements, PCs are suppressed by interneurons [28]. In other words, the activation of cerebellar nucleus neurons generated by reduced inhibition from PCs (disinhibition) facilitates the execution of movement. Thus, chained GABA neurons are essential in the formation of cerebellar output signals.
Synaptic Plasticity of AMPAR
2.5. Receptor Trafficking at PF-PC Synapse
- AMPA-type glutamate receptor. It should be acknowledged that receptor trafficking underlies the plasticity of AMPAR, AP2, and clathrin-dependent endocytosis in LTD and NO (nitric oxide)-dependent exocytosis in LTP. VAMP-dependent constitutive exocytosis of AMPAR is reported in rat PC [82], but not found in mouse PC [83]. Further investigation is required to determine the synaptic recycling of AMPAR at PF-PC synapses in various mammalian species, including primates.
- Metabotropic glutamate receptor 1. Constitutive trafficking of mGluR1 is regulated by Homer and Transferrin receptor (TFR). PC ablation of TFR1 inhibits parallel fiber-PC LTD and results in impairment of motor coordination [84].
3. Clinical Autoimmune Background and Pathophysiological Actions of Autoantibodies on the Ion Channels/Related Proteins and Synaptic Machinery
3.1. Overview
- etiologies in which autoimmunity is triggered by other conditions; for example, gluten sensitivity in gluten ataxia (GA), paraneoplastic conditions in paraneoplastic cerebellar degenerations (PCDs), infections in post-infectious cerebellitis (PIC), and Miller Fisher syndrome, and
3.2. Anti-VGCC Antibody
3.2.1. Clinical Profile of Anti-VGCC Ab-Associated CA
3.2.2. Effects of Autoantibodies
3.3. Anti-mGluR1 Antibody
3.3.1. Clinical Profile of Anti-mGluR1 Ab-Associated CA
3.3.2. Effects of Autoantibodies
3.4. Anti-GAD65 Antibody
3.4.1. Clinical Profile of Anti-GAD65 Ab-Associated CA
3.4.2. Effects of Autoantibodies
- anti-GAD65 Ab is associated with type 1 diabetes mellitus (T1DM) and various neurological conditions, such as epilepsy and Stiff-Person syndrome, and
- GAD65 is intracellularly located, implying that autoantibodies do not have access to GAD65.
3.5. Anti-DPPX Antibody
3.5.1. Clinical Profile of Anti-DPPX Ab-Associated CA
3.5.2. Effects of Autoantibodies
3.6. Anti-Caspr2 Antibody
3.6.1. Clinical Profile of Anti-Caspr2 Ab-Associated CA
3.6.2. Effects of Autoantibodies
3.7. Anti-GluRδ Antibody
3.7.1. Clinical Profile of Anti-GluRδ Ab-Associated CA
3.7.2. Effects of Autoantibodies
3.8. Anti-AMPA Receptor Antibody
3.8.1. Clinical Profile of Anti-AMPAR Ab-Associated CA
3.8.2. Effects of Autoantibodies
3.9. Anti-Glycine Receptor Antibody
3.9.1. Clinical Profile of Anti-Glycine Ab-Associated CA
3.9.2. Effects of Autoantibodies
3.10. Anti-GABAA Receptor Antibody
3.10.1. Clinical Profile of Anti-GABAAR Ab-Associated CA
3.10.2. Effects of Autoantibodies
3.11. Anti-GABAB Receptor Antibody
3.11.1. Clinical Profile of Anti-GABAB R Ab-Associated CA
3.11.2. Effects of Autoantibodies
3.12. Anti-LGl1 Antibody
3.12.1. Clinical Profile of Anti-LGI1 Ab-Associated CA
3.12.2. Effects of Autoantibodies
3.13. Other Autoantibodies Lacking Characterization
4. Fundamental Pathophysiology of Autoantibodies Targeting Ion Channel Functions and Synaptic Transmission
4.1. Region-Specific Vulnerability to Autoantibodies
4.2. Physiological Categorization of Autoantibodies
- ion channels and their related proteins both of which determine neural excitability, and
- synaptic transmission-related proteins (Table 4). The latter is further classified into:
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lancaster, E.; Dalmau, J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurol. 2012, 8, 380–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graus, F.; Saiz, A.; Dalmau, J. Antibodies and neuronal autoimmune disorders of the CNS. J. Neurol. 2010, 257, 509–517. [Google Scholar] [CrossRef]
- Lancaster, E.; Martinex-Hernandez, E.; Dalmau, J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011, 77, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Dalmau, J.; Rosenfeld, M.R. Autoimmune encephalitis update. Neuro. Oncol. 2014, 16, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to synaptic receptors and neuronal cell surface protein in autoimmune diseases of the central nervous system. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef]
- Hadjivassiliou, M. Immune-mediated acquired ataxias. Handb. Clin. Neurol. 2012, 103, 189–199. [Google Scholar] [PubMed]
- Mitoma, H.; Adhikari, K.; Aeschlimann, D.; Chattopadhyay, P.; Hadjivassiliou, M.; Hampe, C.S.; Honnorat, J.; Joubert, B.; Kakei, S.; Lee, J.; et al. Consensus Paper: Neuroimmune mechanisms of cerebellar ataxias. Cerebellum 2016, 15, 2313–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitoma, H.; Hadjivassiliou, M.; Honnorat, J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, H.; Manto, M.; Hampe, C.S. Immune-mediated cerebellar ataxias: From bench to bedside. Cerebellum Ataxias 2017, 4, 16. [Google Scholar] [CrossRef] [Green Version]
- Mitoma, H.; Manto, M.; Hampe, C.S. Immune-mediated cerebellar ataxias: Practical guidelines and therapeutic challenges. Curr. Neuropharmacol. 2018. [Google Scholar] [CrossRef]
- Joubert, B.; Rotásky, J.; Honnorat, J. Immune-mediated ataxias. Handb. Clin. Neurol. 2018, 155, 313–332. [Google Scholar] [PubMed]
- Hadjivassiliou, M.; Mitoma, H.; Manto, M. Autoimmune ataxia. In Neuroimmune Diseases; From Cells to the Living Brain; Mitoma, H., Manto, M., Eds.; Springer Nature: Basel, Switzerland, 2019; pp. 599–620. [Google Scholar]
- Mitoma, H.; Manto, M.; Hadjivassiliou, M. Non-paraneoplastic autoimmune cerebellar diseases. In MedLink Neurology; Roos, R.P., Ed.; MedLink Corporation: San Diego, CA, USA, 2019; Available online: www.medlink.com (accessed on 12 July 2020).
- Joubert, B.; Honnorat, J. Nonparaneoplastic autoimmune cerebellar ataxia. Curr. Opin. Neurol. 2019, 32, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B. Medusa-head ataxia’: The expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: Anti-mGluR1, anti-Homer-3, anti-Sj/ITPR1 and anti-CARP VIII. J. Neuroinflamm. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarius, S.; Wildemann, B. ‘Medusa-head ataxia’: The expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: Anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. J. Neuroinflamm. 2015. [Google Scholar] [CrossRef] [Green Version]
- Jarius, S.; Wildemann, B. ‘Medusa-head ataxia’: The expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: Anti-Yo/CDR2, anti-Nb/AP3B2, PCA-2, anti-Tr/DNER, other antibodies, diagnostic pitfalls, summary and outlook. J. Neuroinflamm. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitoma, H.; Ishida, K.; Shizuka-Ikeda, M.; Mizusawa, H. Dual impairment of GABAA- and GABAB-receptor-mediated synaptic responses by autoantibodies to glutamic acid decarboxylase. J. Neurol. Sci. 2003, 208, 51–56. [Google Scholar] [CrossRef]
- Eccles, J.C.; Ito, M.; Szentagothai, J. The Cerebellum as a Neuronal Machine; Springer: New York, NY, USA, 1967. [Google Scholar]
- Ito, M. The Cerebellum and Neural Control; Raven Press: New York, NY, USA, 1984. [Google Scholar]
- Napper, R.M.; Harvey, R.J. Number of parallel fiber synapses on an individual Purkinje cell in the cerebellum of the rat. J. Comp. Neurol. 1988, 274, 168–177. [Google Scholar] [CrossRef]
- Eccles, J.C.; Llinás, R.; Sasaki, K. The inhibitory interneurones within the cerebellar cortex. Exp. Brain Res. 1966, 1, 1–16. [Google Scholar] [CrossRef]
- Ito, M.; Yoshida, M. The cerebellar-evoked monosynaptic inhibition of Deiters’ neurons. Experimentia 1964, 20, 515–516. [Google Scholar] [CrossRef]
- Manto, M.; Bower, J.M.; Conforto, A.B.; Delgado-García, J.M.; da Guarda, S.N.; Gerwig, M.; Habas, C.; Hagura, N.; Ivry, R.B.; Mariën, P.; et al. Consensus Paper: Roles of the cerebellum in motor control—The diversity of ideas on cerebellar involvement in movement. Cerebellum 2012, 11, 457–847. [Google Scholar] [CrossRef]
- Schmahmann, J.D.; Caplan, D. Cognitive, emotion and the cerebellum. Brain 2006, 129, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Ito, M. Cerebellar long-term depression: Characterization, signal transduction, and functional roles. Physiol. Rev. 2001, 81, 1143–1195. [Google Scholar] [CrossRef]
- Popa, L.S.; Hewitt, A.L.; Ebner, T.J. Predictive and feedback performance errors are signaled in the simple spike discharge of individual Purkinje cells. J. Neurosci. 2012, 32, 15345–15358. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Ishikawa, T.; Lee, J.; Kakei, S. The cerebro-cerebellum as a locus of foraward model: A review. Front. Syst. Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afshari, F.S.; Ptak, K.; Khaliq, Z.M.; Grieco, T.M.; Slater, N.T.; McCrimmon, D.R.; Raman, I.M. Resurgent Na currents in four classes of neurons of the cerebellum. J. Neurophysiol. 2004, 92, 2831–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoli, S.; Solinas, S.; D’Angelo, E. Action potential processing in a detailed Purkinje cell model reveals a critical role for axonal compartmentalization. Front. Cell Neurosci. 2015, 9, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, B.E.; Molineux, M.L.; Mehaffey, W.H.; Turner, R.W. Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. J. Neurosci. 2005, 25, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Zagha, E.; Lang, E.J.; Rudy, B. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J. Neurosci. 2008, 28, 1291–1300. [Google Scholar] [CrossRef]
- Wang, Y.T.; Linden, D.J. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 2000, 25, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Lujan, R.; Aguado, C.; Ciruela, F.; Arus, X.M.; Martin-Belmonte, A.; Alfaro-Ruiz, R.; Martinez-Gomez, J.; de la Ossa, L.; Watanabe, M.; Adelman, J.P.; et al. SK2 channel associate with mGlu1a receptors and CaV2.1 channels in Purkinje cells. Front. Cell Neurosci. 2018, 12, 311. [Google Scholar] [CrossRef] [Green Version]
- Vigot, R.; Batini, C. GABAB receptor activation of Purkinje cells in cerebellar slices. Neurosci. Res. 1997, 29, 151–160. [Google Scholar] [CrossRef]
- Kole, M.J.; Qian, J.; Waase, M.P.; Klassen, T.L.; Chen, T.T.; Augustine, G.J.; Noebels, J.L. Selective loss of presynaptic potassium channel clusters at the cerebellar basket cell terminal Pinceau in Adam11 mutants reveals their role in ephaptic control of Purkinje cell firing. J. Neurosci. 2015, 35, 11433–11444. [Google Scholar] [CrossRef] [Green Version]
- Shibata, R.; Nakahira, K.; Shibasaki, K.; Wakazono, Y.; Imoto, K.; Ikenaka, K. A-Type K+ current mediated by the Kv4 channel regulates the generation of action potential in developing cerebellar granule cells. J. Neurosci. 2000, 20, 4145–4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, K.; Wakamori, M.; Rhyu, I.J.; Arii, T.; Oda, S.; Mori, Y.; Imoto, K. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J. Neurosci. 2002, 22, 4388–4398. [Google Scholar] [CrossRef] [PubMed]
- Maejima, T.; Wollenweber, P.; Teusner, L.U.; Noebels, J.L.; Herlitze, S.; Mark, M.D. Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to Purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J. Neurosci. 2013, 33, 5162–5174. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Ann. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- Stephens, G.J.; Morris, N.P.; Fyffe, R.E.; Robertson, B. The CaV2.1/a1A(P/Q-type) voltage-dependent calcium channel mediates inhibitory neurotransmission onto mouse cerebellar Purkinje cells. Eur. J. Neurosci. 2001, 13, 1902–1912. [Google Scholar] [CrossRef]
- Isope, P.; Hildebrand, M.E.; Snutch, T.P. Contributions of T-type voltage-gated calcium channels to postsynaptic calcium signaling within Purkinje neurons. Cerebellum 2012, 11, 651–665. [Google Scholar] [CrossRef] [Green Version]
- Linás, R.; Sugimori, M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J. Physiol. 1980, 305, 197–213. [Google Scholar] [CrossRef]
- Ito, M. The molecular organization of cerebellar long-term depression. Nature Rev. Neurosci. 2002, 3, 896–902. [Google Scholar] [CrossRef]
- Kano, M.; Rexhausen, U.; Dreessen, J.; Konnerth, A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje calls. Nature 1992, 356, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Maejima, T.; Hashimoto, K.; Yoshida, T.; Aiba, A. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 2001, 31, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Guroli, D.; Manto, M.; Haines, D. Ca2+ signaling in cerebellar Purkinje neurons-Editorial. Cerebellum 2012, 11, 605–608. [Google Scholar]
- Hartman, J.; Dragicevic, E.; Dragicevic, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.G.; Ahn, S.J.; Hong, Y.W.; Chang, W.S.; Kim, J.; Kim, S.J. Transient receptor potential canonical channels regulate the induction of cerebellar long-term depression. J. Neurosci. 2012, 32, 12909–12914. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Karl, R.M.; Alexander, R.P.D.; Adelsberger, H.; Brill, M.S.; Rühlmann, S.; Ansel, A.; Sakimura, K.; Baba, Y.; Kurosaki, T.; et al. STIM1 controls neuronal Ca2+ signaling, mGluR1-dependent synaptic transmission and cerebellar motor behavior. Neuron 2014, 82, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. Neurotransmitter release: The last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Pang, Z.P.; Südhof, T.C. Cell biology of Ca2+-triggered exocytosis. Curr. Opin. Cell Biol. 2010, 22, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Takamori, M.; Hamada, T.; Komai, K.; Takahashi, M.; Yoshida, A. Synaptotagmin can cause an immune-mediated model of Lambert-Eaton myasthenic syndrome in rats. Ann. Neurol. 1994, 35, 74–80. [Google Scholar] [CrossRef]
- Yuzaki, M. Two classes of secreted synaptic organizers in the central nervous system. Ann. Rev. Physiol. 2018, 80, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 is a ligand for an orphan glutamate receptor delta 2, a bidirectional synapse organizer. Science 2010, 328, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Ito-Ishida, A.; Okabe, S.; Yuzaki, M. The role of Cbln1 on Purkinje cell synapse formation. Neurosci. Res. 2014, 83, 64–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Chen, L.Y.; Liu, X.; Maxeiner, S.; Lee, S.J.; Gokce, O.; Südhof, T.C. Neuroligins sculpt cerebellar Purkinje cell circuits by differential control of distinct classes of synapses. Neuron 2015, 87, 781–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredt, D.S.; Nicoll, R.A. AMPA receptor trafficking at excitatory synapses. Neuron 2003, 40, 361–379. [Google Scholar] [CrossRef] [Green Version]
- Brorson, J.R.; Zhang, Z.; Vandenberghe, W. Ca2+ permeation of AMPA receptors in cerebellar neurons expressing Glu receptor 2. J. Neurosci. 1999, 19, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.; Hideyama, T.; Yamashita, T.; Aizawa, H. AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 2010, 30, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Chung, H.J.; Wihler, C.; Huganir, R.L.; Linden, D.J. Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron 2000, 28, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, M.; Le Pichon, C.E.; Jackson, A.C.; Cerpas, M.; Sakinuma, K.; Scearce-Levie, K.; Nicoll, R.A. Relative contribution of TARPs γ-2 and γ-7 to cerebellar excitatory synaptic transmission and motor behavior. Proc. Natl. Acad. Sci. USA 2015, 112, E371–E379. [Google Scholar] [CrossRef] [Green Version]
- Takamiya, K.; Mao, L.; Huganir, R.L.; Linden, D.J. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J. Neurosci. 2008, 28, 5752–5755. [Google Scholar] [CrossRef]
- Galliano, E.; Schonewille, M.; Peter, S.; Rutteman, M.; Houtman, S.; Jaarsma, D.; Hoebeek, F.E.; De Zeeuw, C.I. Impact of NMDA receptor overexpression on cerebellar Purkinje cell activity and motor learning. eNeuro 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Kono, M.; Kakegawa, W.; Yoshida, K.; Yuzaki, M. Interneuronal NMDA receptors regulate long-term depression and motor learning in the cerebellum. J. Physiol. 2019, 597, 903–920. [Google Scholar] [CrossRef] [Green Version]
- Lujan, R.; Roberts, J.D.; Sgigemoto, R.; Ohishi, H.; Somogyi, P. Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J. Chem. Neuroanat. 1997, 13, 219–241. [Google Scholar] [CrossRef]
- Armbrust, K.R.; Wang, X.; Hathorn, T.J.; Cramer, S.W.; Chen, G.; Zu, T.; Kangas, T.; Zink, A.N.; Oz, G.; Ebner, T.J.; et al. Mutant β-III spectrin causes mGluR1α mislocalization and functional deficits in a mouse model of spinocerebellar ataxia type 5. J. Neurosci. 2014, 34, 9891–9904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiba, A.; Kano, M.; Chen, C.; Stanton, M.E.; Fox, G.D.; Herrup, K.; Zwingman, T.A.; Tonegawa, S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell 1994, 79, 377–388. [Google Scholar] [PubMed]
- Briatore, F.; Patrizi, A.; Viltono, L.; Sassoe-Pognetto, M.; Wulff, P. Quantitative organization of GABAergic synapses in the molecular layer of the mouse cerebellar cortex. PLoS ONE 2010, 5, e12119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luján, R.; Aguado, C.; Ciruela, J.; Kleindienst, D.; de la Ossa, L.; Bettler, B.; Wickman, K.; Watanabe, M.; Shigemoto, R.; Fukazwa, Y. Differential association of GABAB receptors with their effector ion channels in Purkinje cells. Brain Struct. Funct. 2018, 223, 1565–1587. [Google Scholar] [CrossRef] [Green Version]
- Schonewille, M.; Gao, Z.; Boele, H.J.; Veloz, M.F.; Amerika, W.E.; Simek, A.A.; De Jeu, M.T.; Steinberg, J.P.; Takamiyam, K.; Hoebeek, F.E.; et al. Reevaluating the role of LTD in cerebellar motor learning. Neuron 2011, 70, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Sakurai, M.; Tongroach, P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol. 1982, 324, 113–134. [Google Scholar] [CrossRef]
- Hirano, T.; Ohmori, H. Voltage-gated and synaptic currents in rat Purkinje cells in dissociated cell cultures. Proc. Natl. Acad. Sci. USA 1986, 83, 1945–1949. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro guinea-pig cerebellar slices. J. Physiol. 1987, 394, 463–480. [Google Scholar] [CrossRef] [PubMed]
- Linden, D.J.; Connor, J.A. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science 1991, 254, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Denk, W.; Hausser, M. Coincidence detection in single dendritic spines mediated by calcium release. Nat. Neurosci. 2000, 3, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamaguchi, K.; Nagao, S.; Yamazaki, T. Long-term depression as a model of cerebellar plasticity. Prog. Brain Res. 2014, 210, 1–30. [Google Scholar]
- Myoga, M.H.; Regehr, W.G. Calcium microdomains near R-type calcium channels control the induction of presynaptic long-term potentiation at parallel fiber to Purkinje cell synapses. J. Neurosci. 2011, 31, 5235–5243. [Google Scholar] [CrossRef] [Green Version]
- Kakegawa, W.; Yuzaki, M. A mechanism underlying AMPA receptor trafficking during cerebellar long-term potentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 17846–17851. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Castellanos, N.; Da Silva-Matos, C.M.; Zhou, K.; Canto, C.B.; Renner, M.C.; Koene, L.M.C.; Ozyildirim, O.; Sprengel, R.; Kessels, H.W.; De Zeeuw, C.I. Motor learning requires Purkinje cell synaptic potentiation through activation of AMPA-receptor subunit GluA3. Neuron 2017, 93, 409–424. [Google Scholar] [CrossRef] [Green Version]
- Tatsukawa, T.; Chimura, T.; Miyakawa, H.; Yamaguchi, K. Involvement of basal protein kinase C and extracellular signal-regulated kinase 1/2 activities in constitutive internalization of AMPA receptors in cerebellar Purkinje cells. J. Neurosci. 2006, 4820–4825. [Google Scholar] [CrossRef]
- Kakegawa, W.; Katoh, A.; Narumi, S.; Miura, E.; Motohashi, J.; Takahashi, A.; Kohda, K.; Fukazawa, Y.; Yuzaki, M.; Matsuda, S. Optogenetic control of synaptic AMPA receptor endocytosis reveals roles of LTD in motor learning. Neuron 2018, 99, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.H.; Wang, X.T.; Zhou, L.; Zhou, L.; Su, L.D.; Wang, H.; Jia, F.; Xu, F.Q.; Chen, G.Q.; De Zeeuw, C.I.; et al. Ablation of TFR1 in Purkinje cells inhibits mGlu1 trafficking and impairs motor coordination, but not autistic-like behaviors. J. Neurosci. 2017, 37, 11335–11352. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Shams’ili, S.; Grefkens, J.; de Leeuw, B.; van den Bent, M.; Hooijkaas, H.; van der Holt, B.; Vecht, C.; Sillevis Smitt, P. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: Analysis of 50 patients. Brain 2003, 126, 1409–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouston, P.D.; Saper, C.B.; Arbizu, T.; Johnston, I.; Lang, B.; Newsom-Davis, J.; Posner, J.B. Paraneoplastic cerebellar degeneration. III. Cerebellar degeneration, cancer, and the Lambert-Eaton myasthenic syndrome. Neurology 1992, 42, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Okamura, T.; Takahashi, H.; Horiguchi, N.; Sunaga, N.; Hisada, T.; Yamada, T. Small-cell lung cancer with voltage-gated calcium channel antibody-positive paraneoplastic limbic encephalitis: A case report. J. Med. Case Rep. 2014, 8, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, W.P.; Graus, F.; Lang, B.; Honnorat, J.; Delattre, J.Y.; Valldeoriola, F.; Antoine, J.C.; Rosenblum, M.K.; Rosenfeld, M.R.; Newsom-Davis, J.; et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain 1997, 120, 1279–1300. [Google Scholar] [CrossRef]
- Graus, F.; Lang, B.; Pozo-Rosich, P.; Saiz, A.; Casamitjana, R.; Vincent, A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002, 59, 764–766. [Google Scholar] [CrossRef]
- Burk, K.; Wick, M.; Roth, G.; Decker, P.; Voltz, R. Antineuronal antibodies in sporadic late-onset cerebellar ataxia. J. Neurol. 2010, 257, 59–62. [Google Scholar] [CrossRef]
- Liao, Y.J.; Safa, P.; Chen, Y.R.; Sobel, R.A.; Boyden, E.S.; Tsien, R.W. Anti-Ca2+ channel antibody attenuates Ca2+ currents and mimics cerebellar ataxia in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2705–2710. [Google Scholar] [CrossRef] [Green Version]
- Martin-Garcia, E.; Mannara, F.; Gutierrez-Cuesta, J.; Sabater, L.; Dalmau, J.; Maldonado, R.; Graus, F. Intrathecal injection of P/Q type voltage-gated calcium channel antibodies from paraneoplastic cerebellar degeneration cause ataxia in mice. J. Neuroimmunol. 2013, 261, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.; Gillard, S.; Moss, F.; Whyte, K.; Brust, P.; Williams, M.; Stauderman, K.; Harpold, M.; Lang, B.; Newsom-Davis, J.; et al. Human autoantibodies specific for the alpha1A calcium channel subunit reduce both P-type and Q-type calcium currents in cerebellar neurons. Proc. Nat. Acad. Sci. USA 1998, 95, 8328–8333. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Motomura, M.; Nakao, Y.; Shiraishi, H.; Yoshimura, T.; Iwanaga, K.; Tsujihata, M.; Eguchi, K. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann. Neurol. 2003, 53, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Sillevis Smitt, P.; Kinoshita, A.; De Leeuw, B.; Moll, W.; Coesmans, M.; Jaarsma, D.; Henzen-Logmans, S.; Vecht, C.; De Zeeuw, C.; Sekiyama, N.; et al. Paraneoplastic cerebellar ataxia due to antibodies toward against a glutamate receptor. N. Engl. J. Med. 2000, 342, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Iorio, R.; Damato, V.; Mirabella, M.; Vita, M.G.; Hulsenboom, E.; Plantone, D.; Bizzarro, A.; Del Grande, A.; Sillevis Smitt, P.A. Cerebellar degeneration associated with mGluR1 autoantibodies as a paraneoplastic manifestation of prostate adenocarcinoma. J. Neuroimmunol. 2013, 263, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Marignier, R.; Chenevier, F.; Rogemond, V.; Sillevis Smitt, P.; Renoux, C.; Cavillon, G.; Androdias, G.; Vukusic, S.; Graus, F.; Honnorat, J.; et al. Metabotropic glutamate receptor type 1 autoantibody-associated cerebellitis: A primary autoimmune disease? Arch. Neurol. 2010, 67, 627–630. [Google Scholar] [CrossRef] [Green Version]
- Coesmans, M.; Smitt, P.A.; Linden, D.J.; Shigemoto, R.; Hirano, T.; Yamakawa, Y.; van Alphen, A.M.; Luo, C.; van der Geest, J.N.; Kros, J.M.; et al. Mechanisms underlying cerebellar motor deficits due to mGluR1-autoantibodies. Ann. Neurol. 2003, 53, 325–336. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, D.; Grünewald, R.A.; Sanders, D.S.; Sharrack, B.; Woodroofe, N. GAD antibody-associated neurological illness and its relationship to gluten sensitivity. Acta Neurol. Scand. 2011, 123, 175–180. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic roles of glutamate decarboxylase 65 autoantibodies in cerebellar ataxias. J. Immunol. Res. 2017, 2913297. [Google Scholar]
- Honnorat, J.; Saiz, A.; Giometto, B.; Vincent, A.; Brieva, L.; Andres, C.; Maestre, J.; Fabien, N.; Vighetto, A.; Casamitjana, R.; et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies. Study of 14 patients. Arch. Neurol. 2001, 58, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Arińo, H.; Gresa –Arribas, N.; Blanco, Y.; Martínez-Hernández, E.; Sabater, L.; Petit-Pedrol, M.; Rouco, I.; Bataller, L.; Dalmau, J.O.; Saiz, A.; et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies. Immunologic profile and long-term effect of immunotherapy. JAMA Neurol. 2014, 71, 1009–10016. [Google Scholar] [CrossRef] [Green Version]
- Manto, M.; Mitoma, H.; Hampe, C.S. Anti-GAD antibodies and the cerebellum: Where do we stand? Cerebellum 2018, 18, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Gresa-Arribas, N.; Arińo, H.; Martínez-Hernández, F.; Petit-Pedrol, M.; Sabater, L.; Saiz, A.; Dalmau, J.; Graus, F. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS ONE 2015, 10, e0121364. [Google Scholar] [CrossRef] [PubMed]
- Balint, B.; Bhatia, K.P. Stiff person syndrome and other immune-mediated movement disorders—New insights. Curr. Opin. Neurol. 2016, 29, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Mitoma, H.; Song, S.Y.; Uchihara, T.; Inaba, A.; Eguchi, S.; Kobayashi, T.; Mizusawa, H. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann. Neurol. 1999, 46, 263–267. [Google Scholar] [CrossRef]
- Mitoma, H.; Song, S.Y.; Ishida, K.; Yamakuni, T.; Kobayashi, T.; Mizusawa, H. Presynaptic impairment of cerebellar inhibitory synapses by an autoantibody to glutamic acid decarboxylase. J. Neurol. Sci. 2000, 175, 40–44. [Google Scholar] [CrossRef]
- Manto, M.U.; Laute, M.A.; Aguera, M.; Rogemond, V.; Pandolfo, M.; Honnorat, J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann. Neurol. 2007, 61, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Manto, M.U.; Hampe, C.S.; Rogemond, V.; Honnorat, J. Respective implications of glutamate decarboxylase antibodies in stiff person syndrome and cerebellar ataxia. Orphanet J. Rare Dis. 2011, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Manto, M.; Honnorat, J.; Hampe, C.S.; Guerra-Narbona, R.; López-Ramos, J.C.; Delgado-García, J.M.; Saitow, F.; Suzuki, H.; Yanagawa, Y.; Mizusawa, H.; et al. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission. Front. Behav. Neurosci. 2015, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Ishida, K.; Mitoma, H.; Mizusawa, H. Reversibility of cerebellar GABAergic synapse impairment induced by anti-glutamic acid decarboxylase autoantibodies. J. Neurol. Sci. 2008, 271, 186–190. [Google Scholar] [CrossRef]
- Hampe, C.S.; Petrosini, L.; De Bartolo, P.; Caporali, P.; Cutuli, D.; Laricchiuta, D.; Foti, F.; Radtke, J.R.; Vidova, V.; Honnorat, J.; et al. Monoclonal antibodies to 65kDa glutamate decarboxylase induce epitope specific effects on motor and cognitive functions in rats. Orphanet J. Rare Dis. 2013, 8, 8. [Google Scholar] [CrossRef]
- Ishida, K.; Mitoma, H.; Wada, Y.; Oka, T.; Shibahara, J.; Saito, Y.; Murayama, S.; Mizusawa, H. Selective loss of Purkinje cells in a patient with anti-glutamic acid decarboxylase antibody-associated cerebellar ataxia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 190–192. [Google Scholar] [CrossRef] [Green Version]
- Boronat, A.; Gelfand, J.M.; Gresha-Arribas, N.; Jeong, H.-Y.; Walsh, M.; Roberts, K.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R.; Graus, F.; et al. Encephalitis and antibodies to DPPX, a subunit of Kv4.2 potassium channels. Ann. Neurol. 2013, 73, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; Sandroni, P.; et al. DPPX potassium channel antibody; frequency, clinical accompaniments and outcomes in 20 patients. Neurology 2014, 83, 1797–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balint, B.; Jarius, S.; Nagel, S.; Haberkorn, U.; Probst, C.; Blöcker, I.M.; Bahtz, R.; Komorowski, L.; Stöcker, W.; Kastrup, A.; et al. Progressive encephalomyelitis with rigidity and myoclonus: A new variant with DPPX antibodies. Neurology 2014, 82, 1521–1528. [Google Scholar] [CrossRef]
- Kaulin, Y.A.; De Santiago-Castillo, J.A.; Rocha, C.A.; Nadal, M.S.; Rudy, B.; Covarrubias, M. The dipeptidyl-peptidase-like protein DPP6 determines the unitary conductance of neuronal Kv4. 2 channels. J. Neurosci. 2009, 29, 3242–3251. [Google Scholar] [CrossRef]
- Van Sonderen, A.; Ariño, H.; Petit-Pedrol, M.; Leypoldt, F.; Körtvélyessy, P.; Wandinger, K.P.; Lancaster, E.; Wirtz, P.W.; Schreurs, M.W.; Sillevis Smitt, P.A.; et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016, 87, 521–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, B.; Gobert, F.; Thomas, L.; Saint-Martin, M.; Desestret, V.; Convers, P.; Rogemond, V.; Picard, G.; Ducray, F.; Psimaras, D.; et al. Autoimmune episodic ataxia with anti-CASPR2 antibody-associated encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2017, 14, e371. [Google Scholar] [CrossRef] [Green Version]
- Muñiz-Castrillo, S.; Joubert, B.; Elsensohn, M.-H.; Anne-Laurie Pinto, A.-L.; Saint-Martin, M.; Vogrig, A.; Picard, G.; Rogemond, V.; Dubois, V.; Tamouza, R.; et al. Anti-CASPR2 clinical phenotypes correlate with HLA and immunological features. J. Neurol. Neurosurg. Psychiatry 2020, in press. [Google Scholar]
- Saint-Martin, M.; Pieters, A.; Déchelotte, B.; Malleval, C.; Pinatel, D.; Pascual, O.; Karagogeos, D.; Honnorat, J.; Pellier-Monnin, V.; Noraz, N. Impact of anti-CASPR2 autoantibodies from patients with autoimmune encephalitis on CASPR2/TAG-1 interaction and Kv1 expression. J. Autoimmun. 2019, 103, 102284. [Google Scholar] [CrossRef]
- Patterson, K.R.; Dalmalu, J.; Lancaster, E. Mechanisms of Caspr2 antibodies in autoimmune encephalitis and neuromyotonia. Ann. Neurol. 2018, 83, 40–51. [Google Scholar] [CrossRef]
- Sugiyama, N.; Hamano, S.; Mochizuki, M.; Tanaka, M.; Takahashi, Y. A case of chronic cerebellitis with anti-glutamate receptor delta 2 antibody. No To Hattatsu 2004, 36, 60–63. [Google Scholar]
- Shimokaze, T.; Kato, M.; Yoshimura, Y.; Takahashi, Y.; Hayasaka, K. A case of acute cerebellitis accompanied by autoantibodies against glutamate receptor delta2. Brain Dev. 2007, 29, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Shiihara, T.; Kato, M.; Konno, A.; Takahashi, Y.; Hayasaka, K. Acute cerebellar ataxia and consecutive cerebellitis produced by glutamate receptor delta2 autoantibody. Brain Dev. 2007, 29, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Usui, D.; Mitsuda, N.; Hosokawa, T.; Fujieda, M.; Takahashi, Y.; Wakiguchi, H. A case of persistent cerebellar ataxia complicated by conversion disorder—Confirmed by positive cerebrospinal fluid glutamate receptor delta2 and epsilon2 antibodies. No To Hattatsu 2011, 43, 41–45. [Google Scholar]
- Ichikawa, K.; Kikuchi, M.; Takeshita, S.; Nezu, A. A case of chronic recurrent cerebellar ataxia responding to steroid therapy. Brain Dev. 2009, 31, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Launey, T.; Mikawa, S.; Torashima, T.; Yanagihara, D.; Kasaura, T.; Miyamoto, A.; Yuzaki, M. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat. Neurosci. 2003, 6, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Panzer, J.A.; Gleichman, A.J.; Lynch, D.R. Glutamatergic autoencephalitides: An emerging field. J. Neural. Transm. 2014, 121, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Hughes, E.G.; Peng, X.; Zhou, L.; Gleichman, A.J.; Shu, H.; Matà, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef]
- Graus, F.; Boronat, A.; Xifró, X.; Boix, M.; Svigelj, V.; García, A.; Palomino, A.; Sabater, L.; Alberch, J.; Saiz, A. The expanding profile of anti-AMPA receptor encephalitis. Neurology 2010, 74, 857–859. [Google Scholar] [CrossRef]
- Höftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; et al. Encephalitis and AMPA receptor antibodies. Neurology 2015, 84, 2403–2412. [Google Scholar] [CrossRef] [Green Version]
- Spatola, M.; Stojanova, V.; Prior, J.O.; Dalmau, J.; Rossetti, A.O. Serial brain 18FDG-PET in anti-AMPA receptor limbic encephalitis. J. Neuroimmunol. 2014, 271, 53–55. [Google Scholar] [CrossRef]
- Gleichman, A.J.; Panzer, J.A.; Baumann, B.H.; Dalmau, J.; Lynch, D.R. Antigenic and mechanistic characterization of anti-AMPA receptor encephalitis. Ann. Clin. Transl. Neurol. 2014, 1, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, H.; Mannara, F.; Werner, C.; Planagumà, J.; Miguez-Cabello, F.; Schmidl, L.; Grünewald, B.; Petit-Pedrol, M.; Kirmse, K.; Classen, J.; et al. Human antibodies against the AMPA receptor subunit GluA2 induce receptor reorganization and memory dysfunction. Neuron 2018, 100, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Swayne, A.; Tjoa, L.; Broadley, S.; Dionisio, S.; Gillis, D.; Jacobson, L.; Woodhall, M.R.; McNabb, A.; Schweitzer, D.; Tsang, B.; et al. Antiglycine receptor antibody related diseases: A case series and literature review. Eur. J. Neurol. 2018, 25, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.; Farrant, M.; Cull-Candy, S.G. Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J. Physiol. 1995, 485, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, M.; Waters, P.; McHugh, J.; Gorman, G.; O’Riordan, S.; Connolly, S.; Hager, H.; Yu, P.; Becker, C.M.; Vincent, A. Progressive encephalomyelitis, rigidity, and myoclonus: A novel glycine receptor antibody. Neurology 2008, 71, 1291–1292. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137 Pt 8, 2178–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexopoulos, H.; Dalakas, M.C. Immunology of stiff person syndrome and other GAD-associated neurological disorders. Expert Rev. Clin. Immunol. 2013, 9, 1043–1053. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Fujii, M.; Li, M.; Lutfi, B.; Kyhos, J.; McElroy, B. High-dose intravenous immune globulin for stiff-person syndrome. N. Engl. J. Med. 2001, 345, 1870–1876. [Google Scholar] [CrossRef]
- Morales, E.; Tapia, R. Neurotransmitters of the cerebellar glomeruli: Uptake and release of labelled gamma-aminobutyric acid, glycine, serotonin and choline in a purified glomerulus fraction and in granular layer slices. Brain Res. 1987, 420, 11–21. [Google Scholar] [CrossRef]
- Petit-Pedrol, M.; Armangue, T.; Peng, X.; Bataller, L.; Cellucci, T.; Davis, R.; McCracken, L.; Martinez-Hernandez, E.; Mason, W.P.; Kruer, M.C.; et al. Encephalitis with refractory seizures, status epilepticus and antibodies to the GABA(A) receptor: A case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014, 13, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Spatola, M.; Petit-Pedrol, M.; Simabukuro, M.M.; Armangue, T.; Castro, F.J.; Barcelo Artigues, M.I.; Julià Benique, M.R.; Benson, L.; Gorman, M.; Felipe, A.; et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology 2017, 88, 1012–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: Case series and characterization of the antigen. Lancet Neurol. 2010, 9, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Maureille, A.; Fenouil, T.; Joubert, B.; Picard, G.; Rogemond, V.; Pinto, A.L.; Thomas, L.; Ducray, F.; Quadrio, I.; Psimaras, D.; et al. Isolated seizures are a common early feature of paraneoplastic anti-GABAB receptor encephalitis. J. Neurol. 2019, 266, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Steinmeyer, F.; Knobel, A.; Streitberger, K.; Hotter, B.; Horn, S.; Heuer, H.; Schreiber, S.J.; Wilhelm, T.; Trefzer, U.; et al. GABAB receptor antibodies in paraneoplastic cerebellar ataxia. J. Neuroimmunol. 2013, 256, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010, 133, 2734–2748. [Google Scholar] [CrossRef]
- Navarro, V.; Kas, A.; Apartis, E.; Chami, L.; Rogemond, V.; Levy, P.; Psimaras, D.; Habert, M.O.; Baulac, M.; Delattre, J.Y.; et al. Motor cortex and hippocampus are the two main cortical targets in LGI1-antibody encephalitis. Brain 2016, 139, 1079–1093. [Google Scholar] [CrossRef] [Green Version]
- Van Sonderen, A.; Schreurs, M.W.; Wirt, Z.P.W.; Sillevis Smitt, P.A.; Titulaer, M.J. From VGKC to LGI1 and Caspr2 encephalitis: The evolution of a disease entity over time. Autoimmun. Rev. 2016, 15, 970–974. [Google Scholar] [CrossRef] [Green Version]
- Petit-Pedrol, M.; Sell, J.; Planagumà, J.; Mannara, F.; Radosevic, M.; Haselmann, H.; Ceanga, M.; Sabater, L.; Spatola, M.; Soto, D.; et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018, 141, 3144–3159. [Google Scholar] [CrossRef]
- Britton, J. Autoimmune epilepsy. Handb. Clin. Neurol. 2016, 133, 219–245. [Google Scholar]
- Twyman, R.E.; Gahring, L.C.; Spiess, J.; Rogers, S.W. Glutamate receptor antibodies activate a subset of receptors and reveal an agonist binding site. Neuron 1995, 14, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effect of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planagumà, J.; Leypoldt, F.; Mannara, F.; Gutiérrez-Cuesta, J.; Martín-García, E.; Aguilar, E.; Titulaer, M.J.; Petit-Pedrol, M.; Jain, A.; Balice-Gordon, R.; et al. Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 2015, 138, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, M.; Manto, M. Neuroimmune Diseases; From Cells to the Living Brain; Springer Nature: Basel, Switzerland, 2019. [Google Scholar]
- Villaseñor, R.; Kuennecke, B.; Ozmen, L.; Ammann, M.; Kugler, C.; Grüninger, F.; Loetscher, H.; Freskgård, P.O.; Collin, L. Region-specific permeability of the blood-brain barrier upon pericyte loss. L. Cereb. Blood Flow Metab. 2017, 37, 3683–3694. [Google Scholar]
- Orešković, D.; Klarica, M. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 2014, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joubert, B.; Belbezier, A.; Haesebaert, J.; Rheims, S.; Ducray, F.; Picard, G.; Rogemond, V.; Psimaras, D.; Berzero, G.; Desestret, V.; et al. Long-term outcomes in temporal lobe epilepsy with glutamate decarboxylase antibodies. J. Neurol. 2020, 267, 2083–2089. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Nature of Autoantigens | Antigens Targeted | Association of Neoplasm |
|---|---|---|
| Intracellular cascades underlying synaptic plasticity | Sj/ITPR-1 | Rare |
| PKC-γ | Nonsmall cell lung carcinoma | |
| Homer-3 | No reports | |
| Involved in clathrin-dependent endocytosis | Ca/ARGHAP26 | Rare |
| Regulators of exocytosis and dendritic branching | Septin-5 | No reports |
| TRIM9/67 | Lung carcinoma | |
| TRIM46 | Small cell lung carcinoma | |
| Unknown | CARP VIII | Melanoma, Ovary carcinoma |
| Neurochondrin | No reports | |
| Nb/AP3B2 | No reports |
| Type of Autoantibodies | Target Epitope | Autoimmune Limbic Encephalitis | Immune-Mediated Cerebellar Ataxias | Actions |
|---|---|---|---|---|
| Anti-LGI1 | Leucine-rich glioma-inactivated 1 (LGI1), one of the voltage-gated potassium channel (VGKC) Kv1 complex | Common phenotype: Men in their 60’ sNon-paraneoplastic Amnesia, confusion/disorientation, seizures | Rarely associated: 20% of the patients with limbic encephalitis developed motor symptoms including CAs | Internalization of Kv1.1 and AMPAR In vitro; CSF IgGs increased presynaptic release probability and impaired induction of LTP In vivo; CSF IgGs elicited memory loss |
| Anti-Caspr2 | Contactin-associated protein-like 2 (Caspr2), an associated protein of VGKC Kv1 | Common phenotype: Men in their 60’ sParaneoplastic (Thymoma), 20%, non-paraneoplastic Limbic encephalitis is the most common phenotype Morvan syndrome is the second common phenotype cognitive disturbance, epilepsy, peripheral nerve hyperexcitability, neuropathic pain, autonomic dysfunction, insomnia, neuropathic pain, weight loss | Sometimes associated: 35% of the patients with limbic encephalitis or Morvan syndrome developed Cas Some patients with limbic encephalitis showed episodic CAs | Functional blockade In vitro; sera inhibited blocking with contactin-2, an adhesion molecule |
| Anti-DPPX | Dipeptidyl-peptidase-like protein-6 (DPPX), an auxiliary subunit of VGKC Kv4.2 | Common phenotype: Men in their 50’ sNon-paraneoplastic Agitation, delusions, hallucinations, myoclonic jerks, seizures | Sometimes associated: CAs are one of the multifocal neurological symptoms A case with pure CAs and myoclonus was reported | Not examined |
| Anti-VGCC | P/Q-type voltage-gated calcium channel (VGCC) | Not documented | Common phenotype: Paraneoplastic (SCLC) mostly, non-paraneoplastic Pancerebellar ataxias Association of Lambert–Eaton myasthenic syndrome | Functional blockade In vitro; a polyclonal peptide decreased Ca2+ currents and impaired synaptic transmissions In vivo; Serum IgGs induced ataxia |
| Type of Autoantibodies | Target Epitope | Autoimmune Limbic Encephalitis | Immune-Mediated Cerebellar Ataxias | Actions |
|---|---|---|---|---|
| Anti-NMDA-R | -NR1-NR2 unit | Common phenotype: Young women Paraneoplastic (ovarian teratoma), 50% Non-paraneoplastic Psychosis, seizures | Not documented | Internalization of NMDAR In vitro: Patients’ CSF reduced number of NMDAR at synapses In vivo: Patients’ CSF altered memory and behavior |
| Anti-AMPA-R | -GluR1,2,3 unit | Common phenotype: Middle to aged women Paraneoplastic (SCLC, Breast cancer, thymoma), 50% Non-paraneoplastic Behavioral change, memory loss | Occasionally associated 14% of the patients developed CAs. | Internalization of AMPAR In vitro: Patients’ CSF reduced number of AMPAR at synapses, and CSF/serum IgGs decreased peak mEPSC and increased interevent interval In vivo: Anti-GluA2 Ab impaired long-term synaptic plasticity and affected learning and memory |
| Anti-mGluR1 | Not documented | Common phenotype -Paraneoplastic (n = 2), Non-paraneoplastic (n = 1) | Functional blockade In vitro: Serum IgGs blocked the glutamate-stimulated formation of inositol phosphates. Serum IgGs inhibited induction of LTD In vivo: Serum IgGs induced ataxic gait and cerebellar learning | |
| Anti-GluRδ | Not documented | Sometimes associated: -Infection (n = 4) | Internalization of AMPAR In vitro: Ab reduced number of AMPAR at synapses In vivo: Ab induced ataxic symptoms | |
| Anti-GABAAR | -α1 and β3 subunits | Common phenotype: Paraneoplastic (thymoma) 40%, Non-paraneoplastic Seizures, memory and cognitive deficits, behavioral changes, psychosis | Rarely associated | -Internalization of GABAAR In vitro: Patients’ CSF reduced number of GABAA-R at synapses |
| Anti-GABABR | -B1 subunit | Common phenotype: Paraneoplastic (SCLC) 50–80%, Non-paraneoplastic Seizures, confusion memory loss | Rarely associated | Not examined |
| Anti-GAD65 | -GAD65 | Common phenotype: Women, in 60’ sIdiopathic, A few in autoimmune conditions such as paraneoplastic and gluten sensitivity Seizures | Common phenotype: Women, in 60’ sIdiopathic, A few in paraneoplastic and gluten sensitivity Prominent gait ataxia, associated with varying degrees of limb ataxia and nystagmus | Functional blockade In vitro: CSF IgGs reduced GABA release In vivo: CSF IgGs induced ataxic symptoms These actions were elicited by The biding of GAD65 by anti-GAD65 Ab itself The access route is unclear |
| Anti-GlycineR | Not documented | Occasionally associated: 13% of the patients developed CAs. | -Internalization of GlycineR -In vitro; IgGs including anti-GlyR Ab reduced number of GlycineR clusters |
| Category | Autoimmune Targets |
|---|---|
| 1. Ion channels and related proteins | |
| K+ channel | Caspr2 (a VGKC Kv1 associated protein) |
| DPPX (DPP6, a regulatory subunit of VGKC Kv4.2) | |
| Ca2+ channel | VGCC (P/Q-type) |
| 2. Synaptic machinery proteins | |
| 2.1 Release machinery proteins | VGCC (P/Q-type) |
| GAD65 | |
| Amphiphysin | |
| 2.2 Synaptic adhesion/organizing molecules | LGI1 |
| GluRδ | |
| 2.3 Receptors | AMPAR |
| NMDAR | |
| mGluR1 | |
| GABAAR | |
| GABABR | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitoma, H.; Honnorat, J.; Yamaguchi, K.; Manto, M. Fundamental Mechanisms of Autoantibody-Induced Impairments on Ion Channels and Synapses in Immune-Mediated Cerebellar Ataxias. Int. J. Mol. Sci. 2020, 21, 4936. https://doi.org/10.3390/ijms21144936
Mitoma H, Honnorat J, Yamaguchi K, Manto M. Fundamental Mechanisms of Autoantibody-Induced Impairments on Ion Channels and Synapses in Immune-Mediated Cerebellar Ataxias. International Journal of Molecular Sciences. 2020; 21(14):4936. https://doi.org/10.3390/ijms21144936
Chicago/Turabian StyleMitoma, Hiroshi, Jerome Honnorat, Kazuhiko Yamaguchi, and Mario Manto. 2020. "Fundamental Mechanisms of Autoantibody-Induced Impairments on Ion Channels and Synapses in Immune-Mediated Cerebellar Ataxias" International Journal of Molecular Sciences 21, no. 14: 4936. https://doi.org/10.3390/ijms21144936
APA StyleMitoma, H., Honnorat, J., Yamaguchi, K., & Manto, M. (2020). Fundamental Mechanisms of Autoantibody-Induced Impairments on Ion Channels and Synapses in Immune-Mediated Cerebellar Ataxias. International Journal of Molecular Sciences, 21(14), 4936. https://doi.org/10.3390/ijms21144936