Inherited Metabolic Disorders Presenting with Ataxia

Abstract
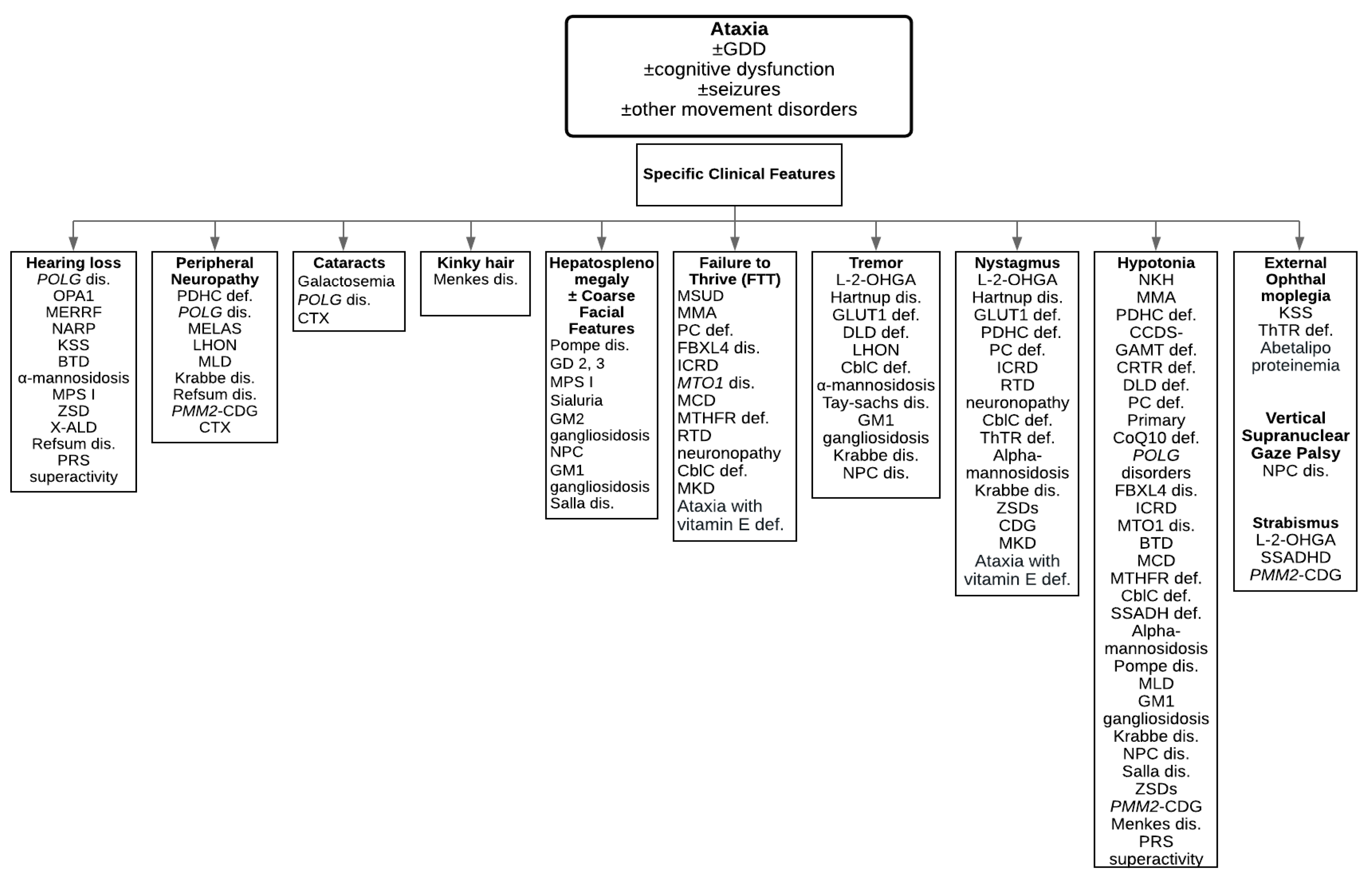
:1. Introduction
2. Treatable Inherited Metabolic Disorders Presenting with Ataxia
2.1. Disorders of Amino Acid Metabolism and Transport
Maple Syrup Urine Disease (MSUD)
2.2. Disorders of Carbohydrate Metabolism
2.2.1. Galactose-1-phosphate Uridylyltransferase Deficiency
2.2.2. Glucose Transporter 1 (GLUT1) Deficiency
2.3. Disorder of Mitochondrial Energy Metabolism
2.3.1. Creatine Deficiency Disorders
2.3.2. Primary Coenzyme Q10 Deficiency
2.4. Vitamin and Cofactor Responsive Disorders
2.4.1. Biotinidase Deficiency
2.4.2. Riboflavin Transporter Deficiency
2.5. Organelle Related Disorders: Lysosomal Storage Disorders
Neuronal Ceroid Lipofuscinosis (NCL)
2.6. Organelle Related Disorders: Peroxisomal Disorders
2.6.1. X-linked adrenoleukodystrophy (X-ALD)
2.6.2. Refsum Disease
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCD1 | ATP-binding cassette domain 1 |
| AGAT | Arginine:glycine amidinotransferase |
| BCKD | Branched-chain alpha-ketoacid dehydrogenase complex |
| BVVL | Brown-Vialetto-Van Laere |
| CDG | Congenital disorders of glycosylation |
| CRTR | Creatine transporter |
| CSF | Cerebrospinal fluid |
| FAD | Flavin adenine dinucleotide |
| FL | Fazio-Londe |
| FMN | Flavin mononucleotide |
| GALT | Galatose-1-phosphate uridylyltransferase |
| GAMT | Guanidinoacetate N-methyltransferase |
| GLUT1 | Glucose transporter 1 |
| HSCT | Hematopoietic stem cell transplantation |
| NCL | Nneuronal ceroid lipofuscinoses |
| PDHC | Pyruvate dehydrogenase complex deficiency |
| MSUD | Maple syrup urine disease |
| X-ALD | X-linked adrenoleukodystrophy |
References
- Poretti, A.; Wolf, N.I.; Boltshauser, E. Differential diagnosis of cerebellar atrophy in childhood. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2008, 12, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Blaser, S.I.; Steinlin, M.; Al-Maawali, A.; Yoon, G. The Pediatric Cerebellum in Inherited Neurodegenerative Disorders: A Pattern-recognition Approach. Neuroimaging Clin. N. Am. 2016, 26, 373–416. [Google Scholar] [CrossRef] [PubMed]
- Akbar, U.; Ashizawa, T. Ataxia. Neurol. Clin. 2015, 33, 225–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashizawa, T.; Xia, G. Ataxia. Contin. Minneap. Minn 2016, 22, 1208–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.R.; Hoffmann, G.F.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. I. Movement disorders. Mol. Genet. Metab. 2019, 127, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, D.; Bullivant, G.; Siriwardena, K.; Evans, A.; Kobayashi, J.; Cohn, R.D.; Mercimek-Andrews, S. Genetic landscape of pediatric movement disorders and management implications. Neurol. Genet. 2018, 4, e265. [Google Scholar] [CrossRef] [Green Version]
- Pode-Shakked, N.; Korman, S.H.; Pode-Shakked, B.; Landau, Y.; Kneller, K.; Abraham, S.; Shaag, A.; Ulanovsky, I.; Daas, S.; Saraf-Levy, T.; et al. Clues and challenges in the diagnosis of intermittent maple syrup urine disease. Eur. J. Med. Genet. 2020, 103901. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Gass, J.M.; Vairo, F.P.E.; Farnham, K.M.; Atwal, H.K.; Macklin, S.; Klee, E.W.; Atwal, P.S. Maple syrup urine disease: Mechanisms and management. Appl. Clin. Genet. 2017, 10, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Strauss, K.A.; Puffenberger, E.G.; Morton, D.H. Maple Syrup Urine Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Neville, S.; O’Sullivan, S.; Sweeney, B.; Lynch, B.; Hanrahan, D.; Knerr, I.; Lynch, S.A.; Crushell, E. Friedreich Ataxia in Classical Galactosaemia. JIMD Rep. 2016, 26, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Berry, G.T. Classic Galactosemia and Clinical Variant Galactosemia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Schweitzer-Krantz, S. Early diagnosis of inherited metabolic disorders towards improving outcome: The controversial issue of galactosaemia. Eur. J. Pediatr. 2003, 162, S50–S53. [Google Scholar] [CrossRef]
- Wang, D.; Pascual, J.M.; De Vivo, D. Glucose Transporter Type 1 Deficiency Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Pons, R.; Collins, A.; Rotstein, M.; Engelstad, K.; De Vivo, D.C. The spectrum of movement disorders in Glut-1 deficiency. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Mercimek-Mahmutoglu, S.; Salomons, G.S. Creatine Deficiency Syndromes. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Khaikin, Y.; Sidky, S.; Abdenur, J.; Anastasi, A.; Ballhausen, D.; Buoni, S.; Chan, A.; Cheillan, D.; Dorison, N.; Goldenberg, A.; et al. Treatment outcome of twenty-two patients with guanidinoacetate methyltransferase deficiency: An international retrospective cohort study. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2018, 22, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.U.; Scaglia, F.; Li, F.-Y.; Smith, L.; Barshop, B.A.; Eng, C.M.; Haas, R.H.; Hunter, J.V.; Lotze, T.; Maranda, B.; et al. Expanded clinical and molecular spectrum of guanidinoacetate methyltransferase (GAMT) deficiency. Mol. Genet. Metab. 2009, 96, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Van de Kamp, J.M.; Betsalel, O.T.; Mercimek-Mahmutoglu, S.; Abulhoul, L.; Grünewald, S.; Anselm, I.; Azzouz, H.; Bratkovic, D.; de Brouwer, A.; Hamel, B.; et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J. Med. Genet. 2013, 50, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Desbats, M.A.; Lunardi, G.; Doimo, M.; Trevisson, E.; Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 2015, 38, 145–156. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hayek, W.; Dumin, Y.; Tal, G.; Zehavi, Y.; Sakran, W.; Spiegel, R. Biotinidase Deficiency: A Treatable Neurological Inborn Error of Metabolism. Isr. Med. Assoc. J. IMAJ 2019, 21, 219–221. [Google Scholar]
- Cowan, T.M.; Blitzer, M.G.; Wolf, B. Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee Technical standards and guidelines for the diagnosis of biotinidase deficiency. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B. Biotinidase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- O’Callaghan, B.; Bosch, A.M.; Houlden, H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J. Inherit. Metab. Dis. 2019, 42, 598–607. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Ejaz, R.; Cordeiro, D.; Kannu, P.; Mercimek-Andrews, S. Riboflavin transporter deficiency mimicking mitochondrial myopathy caused by complex II deficiency. Am. J. Med. Genet. A. 2018, 176, 399–403. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E. Neuronal Ceroid-Lipofuscinoses. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Mole, S.E.; Anderson, G.; Band, H.A.; Berkovic, S.F.; Cooper, J.D.; Kleine Holthaus, S.-M.; McKay, T.R.; Medina, D.L.; Rahim, A.A.; Schulz, A.; et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019, 18, 107–116. [Google Scholar] [CrossRef]
- Schulz, A.; Kohlschütter, A.; Mink, J.; Simonati, A.; Williams, R. NCL diseases-clinical perspectives. Biochim. Biophys. Acta 2013, 1832, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costain, G.; Cordeiro, D.; Matviychuk, D.; Mercimek-Andrews, S. Clinical Application of Targeted Next-Generation Sequencing Panels and Whole Exome Sequencing in Childhood Epilepsy. Neuroscience 2019, 418, 291–310. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.; Ajayi, T.; Specchio, N.; de Los Reyes, E.; Gissen, P.; Ballon, D.; Dyke, J.P.; Cahan, H.; Slasor, P.; Jacoby, D.; et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N. Engl. J. Med. 2018, 378, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Raymond, G.V.; Moser, A.B.; Fatemi, A. X-Linked Adrenoleukodystrophy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Mallack, E.J.; Turk, B.; Yan, H.; Eichler, F.S. The Landscape of Hematopoietic Stem Cell Transplant and Gene Therapy for X-Linked Adrenoleukodystrophy. Curr. Treat. Options Neurol. 2019, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M. Ataxia with Vitamin E Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2005. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1241/ (accessed on 12 June 2020).
- Burnett, J.R.; Hooper, A.J.; Hegele, R.A. Abetalipoproteinemia. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2005. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532447/ (accessed on 12 June 2020).
- Wanders, R.J.A.; Waterham, H.R.; Leroy, B.P. Refsum Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2005. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1353/ (accessed on 12 June 2020).
- Ficicioglu, C.; An Haack, K. Failure to thrive: When to suspect inborn errors of metabolism. Pediatrics. 2009, 124, 972–979. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Category | Disease Name | Gene | Clinical Features (Untreated or No Treatments) |
|---|---|---|---|
| Disorders of amino acid metabolism and transport | Maple syrup urine disease | BCKDHA BCKDHB DBT | GDD, ataxia (episodic or chronic), seizures, FTT, maple syrup odor |
| Nonketotic hyperglycinemia | GLDC AMT | GDD, ataxia, seizures, hypotonia, spasticity | |
| HHH syndrome | SLC25A15 | GDD, cognitive dysfunction, ataxia, spasticity, chronic liver dysfunction, mild or acute encephalopathy | |
| Sulfite oxidase deficiency | SUOX | GDD, movement disorder (episodic or chronic ataxia, dystonia, choreoathetosis), seizures, microcephaly, ectopia lentis | |
| L-2-hydroxyglutaric aciduria | L2HGDH | GDD, speech delay, ataxia, tremor, nystagmus, strabismus, seizures, macrocephaly | |
| Methylmalonic acidemia | MCEE MMADHC | GDD, movement disorder (ataxia, dysarthria), seizures, hypotonia, FTT, intermittent metabolic decompensation, vomiting, lethargy, hepatomegaly, hypothermia | |
| Glutaminase deficiency | GLS | GDD, movement disorder (ataxia, dysarthria), hypertonia | |
| Hartnup disease | SLC6A19 | GDD, movement disorder (ataxia, dystonia, tremor), psychiatric abnormalities, skin rashes, nystagmus | |
| Disorders of carbohydrate metabolism | Galactosemia | GALT | GDD, speech delay, ataxia, liver failure, bleeding, cataracts, premature ovarian failure |
| Glucose transporter 1 deficiency | SLC2A1 | GDD, speech delay, movement disorder (chronic or intermittent ataxia, dysarthria, dystonia, chorea, tremor), nystagmus, seizures, acquired microcephaly | |
| Disorders of mitochondrial energy metabolism | Pyruvate dehydrogenase complex deficiency | PDHA1 PDHB DLAT PDP1 | GDD, intermittent ataxia, nystagmus, seizures, hypotonia, spasticity, microcephaly, peripheral neuropathy, encephalopathy |
| Cerebral creatine deficiency syndromes GAMT deficiency CRTR deficiency | GAMT SLC6A8 | GDD, cognitive dysfunction, speech delay, movement disorder (chronic or episodic ataxia, dystonia, chorea), seizures, behavioural disorder, hypotonia, dysmorphic features (SLC6A8) | |
| Dihydrolipoamide dehydrogenase deficiency | DLD | GDD, ataxia, tremor, seizures, hepatomegaly, liver dysfunction, vision impairment, microcephaly, hypotonia, spasticity | |
| Pyruvate carboxylase deficiency | PC | GDD, ataxia, seizures, hypotonia, FTT, metabolic acidosis, nystagmus | |
| Primary coenzyme Q10 deficiency | COQ2 COQ4 COQ5 COQ6 COQ8A PDSS2 ANO10 | GDD, movement disorder (ataxia, dystonia, parkinsonism), seizures, spasticity, hypotonia, myopathy, encephalopathy, stroke-like episodes, nephrotic syndrome, hypertrophic cardiomyopathy, retinopathy | |
| POLG related disorders | POLG | GDD, movement disorder (ataxia, chorea, parkinsonism), seizures, hypotonia, myopathy, psychiatric illness, stroke-like episodes, peripheral neuropathy, retinopathy, cataracts, hearing loss, liver involvement, endocrine dysfunction, cardiac involvement | |
| Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation | DARS2 | GDD, cognitive dysfunction, motor decline, movement disorder (ataxia, dysarthria), seizures, spasticity | |
| TANGO2 related metabolic encephalopathy and arrhythmias | TANGO2 | GDD, cognitive dysfunction, movement disorders (episodic ataxia, dysarthria), seizures, recurrent acute metabolic crises, rhabdomyolysis | |
| Optic atrophy type 1 | OPA1 | GDD, ataxia, proximal myopathy, visual impairment, vision loss, sensorineural hearing loss | |
| Optic atrophy type 10 | RTN4IP1 | GDD, cognitive dysfunction, ataxia, seizures, low vision | |
| FBXL4 disease | FBXL4 | GDD, ataxia, seizures, lactic acidosis, FTT, hypotonia | |
| MELAS | MT-TL1 MT-ND5 | GDD, ataxia, seizures, stroke-like episodes, recurrent headaches, dementia, lactic acidemia, hearing impairment, peripheral neuropathy, ragged red fibers on muscle biopsy | |
| MERRF | MT-TK MT-TF MT-TL1 MT-TI MT-TP | GDD, movement disorder (ataxia, myoclonus), ragged red fibers on muscle biopsy, lactic acidosis, hearing loss, neuropathy, dementia | |
| Leber hereditary optic neuropathy | MT-ND4 MT-ND6 MT-ND1 | GDD, movement disorder (ataxia, postural tremor), myopathy, vision loss, optic atrophy, peripheral neuropathy | |
| NARP syndrome | MT-ATP6 MT-ND6 | GDD, cognitive dysfunction, ataxia, seizures, muscle weakness, retinopathy, dementia, neuropathy, hearing loss, cardiac conduction defects | |
| Infantile cerebellar-retinal degeneration | ACO2 | GDD, movement disorder (ataxia, athetosis), seizures, FTT, hypotonia, optic atrophy, nystagmus, retinal dystrophy, microcephaly | |
| MNGIE syndrome | TYMP | GDD, ataxia, gastrointestinal dysmobility, cachexia, leukoencephalopathy, sensorimotor neuropathy, ptosis | |
| Kearns–Sayre syndrome | mtDNA deletion | GDD, cognitive dysfunction, ataxia, pigmentary retinopathy, cardiac conduction abnormality, progressive external ophthalmoplegia, hearing loss | |
| MTO1 disease | MTO1 | GDD, ataxia, seizures, FTT, lactic acidosis, hypotonia | |
| Vitamin and cofactor responsive disorders | Biotinidase deficiency | BTD | GDD, ataxia, seizures, hypotonia, skin rash, alopecia, conjunctivitis, hearing loss, vision problems |
| Ataxia with vitamin E deficiency | TTPA | Progressive ataxia, dysdiadochokinesia, dysarthria, macular atrophy, retinitis pigmentosa, nystagmus | |
| Multiple carboxylase deficiency | HLCS | GDD, ataxia, seizures, hypotonia, FTT, vomiting, lethargy, metabolic ketolactic acidosis, skin rash | |
| Methylenetetrahydrofolate reductase deficiency | MTHFR | GDD, cognitive dysfunction, ataxia, seizures, psychiatric symptoms, hypotonia, spasticity, FTT, encephalopathy, microcephaly, apnea, myelopathy | |
| Riboflavin transporter deficiency neuronopathy | SLC52A2 SLC52A3 | GDD, movement disorder (ataxia, tongue fasciculations), nystagmus, muscle weakness, FTT, respiratory insufficiency, nystagmus, sensorineural deafness, optic atrophy | |
| Cobalamin C deficiency | MMACHA | GDD, ataxia, tremor, nystagmus, seizures, hypotonia, FTT, nystagmus, pigmentary retinopathy | |
| Thiamine transporter deficiency | SLC19A2 SLC19A3 | GDD, movement disorders (recurrent ataxia, dystonia, dysarthria), nystagmus, external ophthalmoplegia, seizures, spasticity, eye movement abnormalities, encephalopathy, dysphagia, facial palsy | |
| Neurotransmitter disorders | Succinic semialdehyde dehydrogenase deficiency | ALDH5A1 | GDD, ataxia, seizures, strabismus, behavioural problems, hypotonia |
| Organelle related disorders: lysosomal storage disorders | Neuronal ceroid lipofuscinosis | CLN1 CLN2 CLN5 CLN6 DNAJC5 MFSD8 | GDD, ataxia, seizures, spasticity, blindness, dementia, early death |
| Alpha-mannosidosis | MAN2B1 | GDD, cognitive dysfunction, ataxia, tremor, nystagmus, hypotonia, myopathy, psychiatric symptoms, distinct facial features, skeletal abnormalities, hearing loss, frequent infections | |
| Pompe disease | GAA | GDD, ataxia, hypotonia, hepatomegaly, respiratory insufficiency, cardiomegaly | |
| Fabry disease | GLA | GDD, ataxia, acroparesthesia, angiokeratoma, sweating abnormalities, corneal or lenticular opacity, cardiac disease, renal and cerebrovascular involvement | |
| Metachromatic leukodystrophy | ARSA | GDD, cognitive dysfunction, movement disorders (ataxia, dysarthria), seizures, psychiatric disturbance, hypotonia, spasticity, peripheral neuropathy, gallbladder involvement | |
| Fatty acid hydroxylase-associated neurodegeneration | FA2H | GDD, cognitive dysfunction, movement disorder (ataxia, dystonia, dysarthria), seizures, spasticity, optic atrophy or oculomotor abnormalities | |
| Gaucher disease type 2 Gaucher disease type 3 | GBA | GDD, ataxia, hepatomegaly, splenomegaly, cytopenia, pulmonary involvement, stridor, oculomotor involvement, dysphagia | |
| Multiple sulfatase deficiency | SUMF1 | GDD, ataxia, seizures, spasticity, vertebral abnormalities, skeletal deformities, dental abnormalities, cardiac manifestations, ophthalmic features | |
| Mucopolysaccharidosis type I (Hurler syndrome) | IDUA | GDD, ataxia, coarsened facial features, hepatosplenomegaly, progressive skeletal dysplasia, corneal clouding, hearing loss, cardiac involvement | |
| Sialuria | GNE | GDD, ataxia, neonatal jaundice, hepatomegaly, flat and coarse facial features, microcytic anemia, frequent upper respiratory infections | |
| Tay–Sachs disease | HEXA | GDD, movement disorders (ataxia, dystonia, tremor), seizures, spasticity, increased startle response, vision loss | |
| Sandhoff disease | HEXB | GDD, cognitive dysfunction, ataxia, seizures, spasticity, exaggerated startle response, cherry macules on eyes, splenomegaly, vision loss | |
| GM1 gangliosidosis | GLB1 | GDD, movement disorder (ataxia, dystonia, parkinsonism, tremor), seizures, hypotonia, spasticity, cardiomyopathy, coarsened facial features, skeletal dysplasia | |
| Krabbe disease | GALC | GDD, ataxia, tremor, nystagmus, seizures, behavioural difficulties, hypotonia, spasticity, peripheral neuropathy, vision loss | |
| Sialidosis type I | NEU1 | GDD, ataxia, seizures, cherry red macules, myoclonus, vision loss, corneal opacities | |
| Niemann–Pick type C disease | NPC1 NPC2 | GDD, movement disorder (ataxia, dystonia, dysarthria, tremor, gelastic cataplexy), vertical supranuclear gaze palsy seizures, psychiatric conditions, hypotonia, neonatal jaundice, hepatosplenomegaly, vertical supranuclear gaze palsy, dysphagia | |
| Salla disease | SLC17A5 | GDD, cognitive dysfunction, movement disorder (ataxia, athetosis), seizures, hypotonia, spasticity, facial coarsening | |
| Organelle related disorders: peroxisomal disorders | Zellweger spectrum disorders | PEX2 PEX10 PEX12 PEX16 | GDD, cognitive dysfunction, ataxia, nystagmus, seizures, hypotonia, sensorineural hearing loss, liver dysfunctions, bone stippling, retinal dystrophy |
| X-linked adrenoleukodystrophy | ABCD1 | GDD, ataxia, seizures, behaviour problems, vision loss, hearing loss | |
| Adult refsum disease | PHYH PEX7C | GDD, ataxia, anosmia, retinitis pigmentosa, peripheral neuropathy, hearing loss, ichthyosis, cardiac arrhythmias, skeletal abnormalities | |
| Organelle related disorders: golgi and pre golgi system disorders | PMM2-CDG | PMM2 | GDD, ataxia, nystagmus, strabismus, seizures, hypotonia, peripheral neuropathy, eye, skin, skeletal abnormalities, endocrine dysfunction |
| Disorders of metal transport and metabolism | Aceruloplasminemia | CP | GDD, cognitive dysfunction, movement disorder (ataxia, involuntary movement, dystonia, chorea, dysarthria, parkinsonism), retinal degeneration, diabetes mellitus, anemia |
| Menkes disease | ATP7A | GDD, ataxia, seizures, hypotonia, kinky hair | |
| PKAN | PANK2 | GDD, intellectual impairment, movement disorder (ataxia, dystonia, dysarthria, rigidity, choreoathetosis), spasticity, pigmentary retinal degeneration | |
| PLA2G6 disease | PLA2G6 | GDD, cognitive dysfunction, movement disorder (ataxia in childhood phenotype, dystonia, parkinsonism), psychiatric symptoms (adult phenotype) | |
| Disorders of lipid and bile acid metabolism | Mevalonate kinase deficiency | MVK | GDD, ataxia, nystagmus, FTT, lymphadenopathy, vision problems, hepatosplenomegaly, abdominal pain |
| Abetalipoproteinemia | MTTP | Ataxia, dysarthria, FTT, progressive vision loss, muscle weakness | |
| Cerebrotendinous xanthomatosis | CYP27A1 | GDD, movement disorders (ataxia, dystonia, parkinsonism), seizures, psychiatric disturbances, diarrhea, cataracts, xanthomas, dementia, peripheral neuropathy | |
| Disorders of nucleic acid and heme metabolism | Phosphoribosylpyrophosphate synthetase superactivity | PRPS1 | GDD, cognitive dysfunction, ataxia, hypotonia, hyperuricemia, hyperuricosuria, urinary stone, gouty arthritis, sensorineural hearing loss |
| Purine nucleoside phosphorylase deficiency | PNP | GDD, cognitive dysfunction, ataxia, spasticity, increased risk of autoimmune disorders, recurrent infections |
| Investigations | Type of Investigations | Inherited Metabolic Disorders |
|---|---|---|
| Blood metabolic investigations | Ammonium | MMA |
| Lactate | Mitochondrial disorders | |
| Plasma amino acids | MSUD, NKH, HHH syndrome, glutaminase deficiency | |
| Biotinidase activity | Biotinidase deficiency | |
| Homocysteine | CblC deficiency, MTHFR deficiency | |
| Acylcarnitine profile | MMA | |
| Glucose (paired with CSF glucose) | GLUT1 deficiency | |
| Pyruvate | Pyruvate dehydrogenase complex deficiency, mitochondrial disorders | |
| Enzyme assays for lysosomal storage disorders in WBC | Disease specific enzyme activity measurements | |
| VLCFA | Zellweger spectrum disorders, X-ALD | |
| Transferrine isoelectric focusing | PMM2-CDG | |
| Copper | Menkes disease | |
| Ceruloplasmin | Aceruloplasminemia, Menkes disease | |
| Phytanic acid | Refsum disease | |
| Vitamin E | Ataxia with vitamin E deficiency | |
| LDL-cholesterol, triglyceride, apolipoprotein (apo) B | Abetalipoproteinemia | |
| Galactose-1-phosphate uridylyl transferase activity | Galactosemia | |
| Urine metabolic investigations | Amino acids | Hartnup disease |
| Organic acids | MMA, SSADH deficiency, mevalonate kinase deficiency | |
| Sulfocysteine | SOD | |
| Oligosaccharides | Alpha-mannosidosis | |
| Glycosaminoglycan | MPS | |
| Free and total sialic acid | Salla disease | |
| Guanidinoacetate | GAMT deficiency AGAT deficiency | |
| Creatine to creatinine ratio | Creatine transporter deficiency | |
| CSF | Glucose | GLUT1 deficiency |
| Lactate | GLUT1 deficiency, PDH complex deficiency, mitochondrial disorders | |
| Amino acids | NKH | |
| Neurotransmitters | Inherited neurotransmitter disorders | |
| MTHF | Methylenetetrahydrofolate reductase deficiency | |
| GABA (total and free) | SSADH deficiency | |
| Muscle biopsy | Muscle histology | Mitochondrial disorders |
| Muscle electron microscopy | Mitochondrial disorders | |
| Respiratory chain enzyme activity meausurements | Mitochondrial disorders | |
| Coenzyme Q10 measurement | Co-enzyme Q10 deficiency | |
| Skin fibroblasts | Respiratory chain enzyme activity measurement | Mitochondrial disorders |
| Pyruvate dehydrogenase activity measurement | Pyruvate dehydrogenase complex deficiency | |
| Pyruvate carboxylase activity measurement | Pyruvate carboxylase deficiency | |
| Molecular genetic investigations | Targeted next generation sequencing panel for | Leigh disease, mitochondrial disorders, ataxia |
| Whole exome sequencing | Non-targeted molecular genetic investigation | |
| Mitochondrial genome sequencing | Non-targeted molecular genetic investigation |
| Disease Name | Biochemical Features | Neuroimaging | Treatments |
|---|---|---|---|
| Maple syrup urine disease | ↑ leucine, alloisoleucine, isoleucine, valine in plasma amino acid analysis ↑ ketones and metabolic acidosis during acute metabolic decompensation | Diffusion restriction in cerebellum, WM, BS, BG | Leucine-restricted diet, medical formula, thiamine Branched chain amino acid diet restriction |
| Hartnup disease | ↑ neutral amino acids (alanine, serine, threonine, valine, leucine, isoleucine, phenylalanine, tyrosine, tryptophan, histidine, citrulline, asparagine, glutamine) in urine amino acid analysis | Diffuse brain atrophy | Nicotinamide, neomycin, tryptophan ethyl ester, tryptophan rich protein intake |
| Riboflavin transporter deficiency neuronopathy | Abnormal acylcarnitine profile (elevated short, medium or long chain species) | Normal to cerebellar atrophy, increased T2 intensity in brain stem, cerebellum | Riboflavin |
| Biotinidase deficiency | ↓ Serum biotinidase activity ↑ 3-methylcrotonylglycine, 3-hydroxyisovaleric acid, methylcitrate, 3-hydroxypropionate in urine organic acid analysis Metabolic ketolactic acidosis Hyperammonemia | Cerebral or cerebellar atrophy, delayed myelination | Biotin |
| Multiple carboxylase deficiency | ↑ hydroxypentanoylcarnitine ↑ 3-methylcrotonylglycine, 3-hydroxyisovaleric acid, methylcitrate, 3-hydroxypropionate in urine organic acid analysis Metabolic ketolactic acidosis | Cerebral atrophy, delayed myelination | Biotin |
| Thiamine transporter deficiency | Sometimes ↑ CSF and blood lactate | Atrophy of caudate and putamen, swelling of pons | Biotin, thiamine |
| Methylenetetrahydrofolate reductase deficiency | ↑ plasma homocysteine ↓ to normal methionine in plasma amino acid analysis | Brain atrophy, increased WM signal in T2 | Betaine, folic acid, methionine, pyridoxine, carnitine, 5-methyltetrahydrofolate |
| Cobalamin C deficiency | ↑ plasma homocysteine ↓ to normal methionine in plasma amino acid analysis ↑ methylmalonic acid in urine organic acid analysis | Brain atrophy, WM edema | Hydroxocobalamin, betaine, carnitine, folic acid |
| Galactosemia | ↑ erythrocyte galactose-1-phosphate ↓ erythrocyte GALT activity | Cerebellar and cerebral atrophy, delayed myelination | Galactose and lactose free diet, vitamin D, calcium |
| Glucose transporter 1 deficiency | ↓ CSF glucose with normal blood glucose ↓ erythrocyte 3-O-methyl-D-glucose uptake | Normal | Ketogenic diet |
| Cerebral creatine deficiency syndromes | ↑ urine, plasma GAA (GAMT deficiency) ↑ urine creatine to creatinine ratio | Normal to increased T2 signal in BG | GAMT deficiency: creatinine, ornithine, arginine restricted diet CRTR deficiency: arginine, glycine, creatine |
| Primary coenzyme Q10 deficiency | ↓ coenzyme Q10 in skeletal muscle ↓ complex I+III and II+III activity in muscle | Cerebellar atrophy, and increased T2 signal intensity cerebellum | Coenzyme Q10 |
| Cerebrotendinous xanthomatosis | ↑ cholestanol in plasma ↓ to normal plasma cholesterol | Diffuse brain atrophy, increased signal intensity in WM, substantia nigra, spinal cord in T2 | Chenodeoxycholic acid |
| Niemann-pick type C disease | ↑ oxysterols in plasma Positive filipin staining in cultured fibroblasts | Cerebral and cerebellar atrophy, increased WM intensity in T2 | Miglustat |
| Pyruvate dehydrogenase complex deficiency | ↑ Blood and CSF lactate ↑ Blood and CSF pyruvate and alanine Normal lactate to pyruvate ratio | Cerebral and cerebellar atrophy, increased signal in striatum and thalamus in T2 | Thiamine, carnitine, lipoic acid, ketogenic diet |
| Dihydrolipoamide dehydrogenase deficiency | ↑ Blood and CSF lactate ↑ Blood and CSF pyruvate and alanine ↑ alpha ketoglutarate in urine organic acid analysis ↑ leucine, valine, isoleucine, alloisoleucine in plasma amino acid analysis | Increased signal intensity in BG in T2 | Thiamine, ketogenic diet |
| HHH syndrome | ↑ ammonia ↑ ornithine in plasma amino acid analysis ↑ homocitrulline in urine amino acid analysis | Cerebral atrophy, increased WM signal, increased BG signal, stroke-like lesions | Citrulline, arginine, sodium phenylbutyrate, protein restricted diet |
| Adult refsum disease | ↑ plasma phytanic acid | Normal or cerebral atrophy | Phytanic acid restricted diet |
| Aceruloplasminemia | ↓ serum ceruloplasmin ↓ serum copper or iron ↑ serum ferritin ↑ hepatic iron | Decreased signal intensity in BG in T2 | Iron chelating agents (desferrioxamine, deferiprone, or deferasirox), combined IV desferrioxamine and fresh-frozen human plasma (FFP) |
| Pyruvate carboxylase deficiency | ↑ lactate Normal lactate to pyruvate ratio ↑ alanine, citrulline, lysine in plasma and urine amino acid analysis ↓ aspartic acid, glutamine in plasma and urine amino acid analysis | Hypomyelination, cysts in cortex, BG, brain stem and, cerebellum | Acute management: IV glucose Chronic management: citrate, aspartate, biotin, liver transplantation |
| Alpha-mannosidosis | ↓ alpha-mannosidase activity | Cerebral and cerebellar atrophy | Velmanase alfa (where approved) |
| Fabry disease | ↓ alpha-galactosidase A activity ↑ globotriaosylsphingosine in urine and plasma | Cerebral atrophy, increased signal intensity in WM in T2, stroke-like lesions | Agalsidase beta |
| Neuronal ceroid lipofuscinosis type 2 CLN2 disease | ↓ tripeptidyl peptidase 1 activity | Cerebral and cerebellar atrophy, dark thalami in T2 | Cerliponase alfa intracerebroventricular |
| Mucopolysaccharidosis type I (Hurler syndrome) | ↓ alpha-L-iduronidase activity ↑ urinary glycosaminoglycans ↑ heparan dermatan sulfate in urine glucose amino glucan analysis. | Cerebellar hypoplasia | HSCT Laronidase (for non-CNS manifestations) |
| Krabbe disease | ↓ galactocerebrosidase activity | Cerebral atrophy, demyelination in brain stem and cerebellum, chiasmatic enlargement | HSCT |
| Metachromatic leukodystrophy | ↓arylsulfatatase A activity ↑sulfatides in urine | Cerebral atrophy, demyelination in brain stem and cerebellum, chiasmatic enlargement | HSCT |
| X-linked adrenoleukodystrophy | ↑ VLCFA in plasma | Symmetric enhanced T2 signal in the parieto-occipital region with contrast enhancement at the advancing margin | HSCT |
| Ataxia with vitamin E deficiency | ↓ vitamin E level | Cerebellar atrophy, small T2 high-intensity spots in the periventricular region and the deep white matter | Oral vitamin E supplementation |
| Abetalipoproteinemia | ↓LDL-cholesterol, triglyceride, and apolipoprotein (apo) B | Delayed myelination | Low-fat diet, essential fatty acid supplementation, fat soluable vitamin supplementation (ADEK) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silver, G.; Mercimek-Andrews, S. Inherited Metabolic Disorders Presenting with Ataxia. Int. J. Mol. Sci. 2020, 21, 5519. https://doi.org/10.3390/ijms21155519
Silver G, Mercimek-Andrews S. Inherited Metabolic Disorders Presenting with Ataxia. International Journal of Molecular Sciences. 2020; 21(15):5519. https://doi.org/10.3390/ijms21155519
Chicago/Turabian StyleSilver, Grace, and Saadet Mercimek-Andrews. 2020. "Inherited Metabolic Disorders Presenting with Ataxia" International Journal of Molecular Sciences 21, no. 15: 5519. https://doi.org/10.3390/ijms21155519
APA StyleSilver, G., & Mercimek-Andrews, S. (2020). Inherited Metabolic Disorders Presenting with Ataxia. International Journal of Molecular Sciences, 21(15), 5519. https://doi.org/10.3390/ijms21155519