Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia


 ,
,
Abstract
:1. Introduction
2. Results
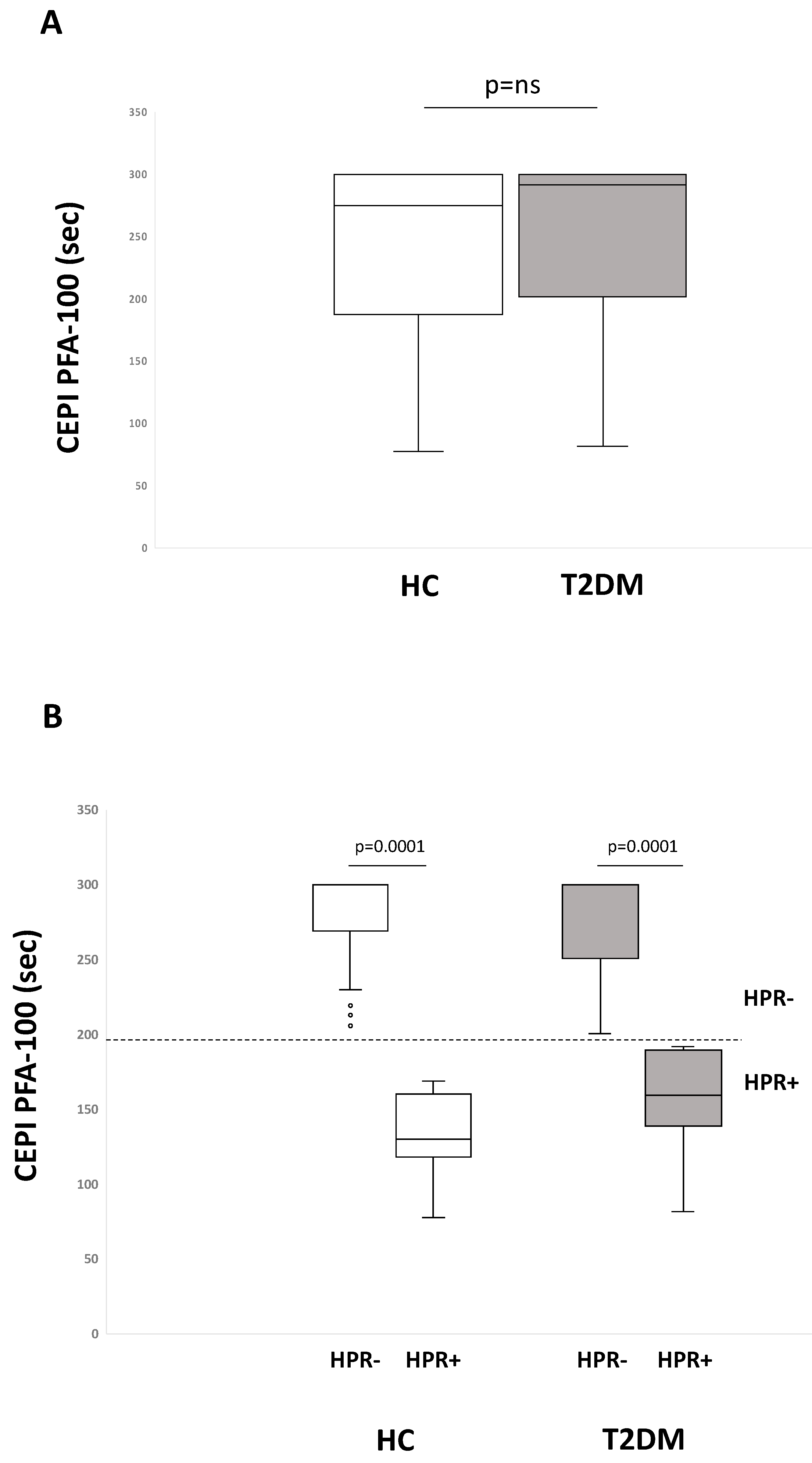
2.1. Residual On-Aspirin Platelet Reactivity Assays in HC and T2DM Patients
2.2. Metabolic Parameters and Biomarkers of Oxidative Stress, Inflammation, and Platelet Activation in HPR+ vs. HPR- according to CEPI PFA-100
2.3. Correlation Analyses
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Laboratory Measurements
4.3. Platelet Assays
4.3.1. PFA-100
4.3.2. Platelet Aggregation
4.3.3. Thromboxane Metabolites
4.4. Biochemical Parameters
4.4.1. Inflammation, Endothelial Dysfunction and in Vivo Platelet Activation
4.4.2. Oxidative Stress
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stratmann, B.; Tschoepe, D. Atherogenesis and atherothrombosis—Focus on diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.P. Diabetes and cardiovascular disease. The “common soil” hypothesis. Diabetes 1995, 44, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Sakkinen, P.A.; Wahl, P.; Cushman, M.; Lewis, M.R.; Tracy, R.P. Clustering of procoagulation, inflammation, and fibrinolysis variables with metabolic factors in insulin resistance syndrome. Am. J. Epidemiol. 2000, 152, 897–907. [Google Scholar] [CrossRef] [Green Version]
- Antithrombotic Trialists’; (ATT) Collaboration; Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; et al. Aspirin in the primary and secondary prevention of vascular disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009, 373, 1849–1860. [Google Scholar]
- Ferreiro, J.L.; Angiolillo, D.J. Diabetes and antiplatelet therapy in acute coronary syndrome. Circulation 2011, 123, 798–813. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Russo, I.; Trovati, M. Resistance to aspirin and thienopyridines in diabetes mellitus and metabolic syndrome. Curr. Vasc. Pharmacol. 2008, 6, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Sacco, M.; Pellegrini, F.; Roncaglioni, M.C.; Avanzini, F.; Tognoni, G.; Nicolucci, A.; PPP Collaborative Group. Primary prevention of cardiovascular events with low-dose aspirin and vitamin E in type 2 diabetic patients: Results of the Primary Prevention Project (PPP) trial. Diabetes Care 2003, 26, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- Russo, I.; Viretto, M.; Barale, C.; Mattiello, L.; Doronzo, G.; Pagliarino, A.; Cavalot, F.; Trovati, M.; Anfossi, G. High glucose inhibits the aspirin-induced activation of the nitric oxide/cGMP/cGMP-dependent protein kinase pathway and does not affect the aspirin-induced inhibition of thromboxane synthesis in human platelets. Diabetes 2012, 61, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Davì, G.; Gresele, P.; Violi, F.; Basili, S.; Catalano, M.; Giammarresi, C.; Volpato, R.; Nenci, G.G.; Ciabattoni, G.; Patrono, C. Diabetes mellitus, hypercholesterolemia, and hypertension but not vascular disease per se are associated with persistent platelet activation in vivo. Evidence derived from the study of peripheral arterial disease. Circulation 1997, 96, 69–75. [Google Scholar] [CrossRef]
- Friend, M.; Vucenik, I.; Miller, M. Research pointers: Platelet responsiveness to aspirin in patients with hyperlipidaemia. BMJ 2003, 326, 82–83. [Google Scholar] [CrossRef] [Green Version]
- Eikelboom, J.W.; Hirsh, J.; Weitz, J.I.; Johnston, M.; Yi, Q.; Yusuf, S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation 2002, 105, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikelboom, J.W.; Hankey, G.J.; Thom, J.; Bhatt, D.L.; Steg, P.G.; Montalescot, G.; Johnston, S.C.; Steinhubl, S.R.; Mak, K.-H.; Easton, J.D.; et al. Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: Determinants and effect on cardiovascular risk. Circulation 2008, 118, 1705–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresele, P.; Guglielmini, G.; De Angelis, M.; Ciferri, S.; Ciofetta, M.; Falcinelli, E.; Lalli, C.; Ciabattoni, G.; Davì, G.; Bolli, G.B. Acute, short-term hyperglycemia enhances shear stress-induced platelet activation in patients with type II diabetes mellitus. J. Am. Coll. Cardiol. 2003, 41, 1013–1020. [Google Scholar] [CrossRef] [Green Version]
- Santilli, F.; Lapenna, D.; La Barba, S.; Davì, G. Oxidative stress-related mechanisms affecting response to aspirin in diabetes mellitus. Free Radic. Biol. Med. 2015, 80, 101–110. [Google Scholar] [CrossRef]
- Davì, G.; Guagnano, M.T.; Ciabattoni, G.; Basili, S.; Falco, A.; Marinopiccoli, M.; Nutini, M.; Sensi, S.; Patrono, C. Platelet activation in obese women: Role of inflammation and oxidant stress. JAMA 2002, 288, 2008–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.A.; Anand, S.S.; Hankey, G.J.; Eikelboom, J.W. Aspirin resistance. Thromb. Res. 2007, 120, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Strålin, P.; Karlsson, K.; Johansson, B.O.; Marklund, S.L. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2032–2036. [Google Scholar] [CrossRef]
- Davì, G.; Ciabattoni, G.; Consoli, A.; Mezzetti, A.; Falco, A.; Santarone, S.; Pennese, E.; Vitacolonna, E.; Bucciarelli, T.; Costantini, F.; et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: Effects of improved metabolic control and vitamin E supplementation. Circulation 1999, 99, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Santilli, F.; Davì, G.; Consoli, A.; Cipollone, F.; Mezzetti, A.; Falco, A.; Taraborelli, T.; Devangelio, E.; Ciabattoni, G.; Basili, S.; et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2006, 47, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Cavalot, F.; Pagliarino, A.; Valle, M.; Di Martino, L.; Bonomo, K.; Massucco, P.; Anfossi, G.; Trovati, M. Postprandial blood glucose predicts cardiovascular events and all-cause mortality in type 2 diabetes in a 14-year follow-up: Lessons from the San Luigi Gonzaga Diabetes Study. Diabetes Care 2011, 34, 2237–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Basili, S.; Falco, A.; Davì, G. Platelet activation in type 2 diabetes mellitus. J. Thromb. Haemost. 2004, 2, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Monnier, L.; Mas, E.; Ginet, C.; Michel, F.; Villon, L.; Cristol, J.-P.; Colette, C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA 2006, 295, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Basili, S.; Davi, G. Platelet activation, inflammatory mediators and hypercholesterolemia. Curr. Vasc. Pharmacol. 2003, 1, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Howard, P.A. Aspirin resistance. Ann. Pharm. 2002, 36, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Barale, C.; Russo, I. Influence of cardiometabolic risk factors on platelet function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrono, C.; Rocca, B. Drug insight: Aspirin resistance--fact or fashion? Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 42–50. [Google Scholar] [CrossRef]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef]
- Gardner, C.D.; Fortmann, S.P.; Krauss, R.M. Association of small low-density lipoprotein particles with the incidence of coronary artery disease in men and women. JAMA 1996, 276, 875–881. [Google Scholar] [CrossRef]
- Harrison, P.; Robinson, M.S.; Mackie, I.J.; Joseph, J.; McDonald, S.J.; Liesner, R.; Savidge, G.F.; Pasi, J.; Machin, S.J. Performance of the platelet function analyser PFA-100 in testing abnormalities of primary haemostasis. Blood Coagul. Fibrinolysis 1999, 10, 25–31. [Google Scholar] [CrossRef]
- Ajjan, R.; Storey, R.F.; Grant, P.J. Aspirin resistance and diabetes mellitus. Diabetologia 2008, 51, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, A.; Zaman, A.G.; Marshall, S.M. Platelet hyperactivity in type 2 diabetes: Role of antiplatelet agents. Diab. Vasc. Dis. Res. 2008, 5, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Rocca, B.; Santilli, F.; Pitocco, D.; Mucci, L.; Petrucci, G.; Vitacolonna, E.; Lattanzio, S.; Mattoscio, D.; Zaccardi, F.; Liani, R.; et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J. Thromb. Haemost. 2012, 10, 1220–1230. [Google Scholar] [CrossRef]
- Santilli, F.; Vazzana, N.; Bucciarelli, L.G.; Davì, G. Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Curr. Med. Chem. 2009, 16, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Dib, M.; Hausding, M.; Kashani, F.; Oelze, M.; Kröller-Schön, S.; Hanf, A.; Daub, S.; Roohani, S.; Gramlich, Y.; et al. CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc. Res. 2018, 114, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- LeLeiko, R.M.; Vaccari, C.S.; Sola, S.; Merchant, N.; Nagamia, S.H.; Thoenes, M.; Khan, B.V. Usefulness of elevations in serum choline and free F2)-isoprostane to predict 30-day cardiovascular outcomes in patients with acute coronary syndrome. Am. J. Cardiol. 2009, 104, 638–643. [Google Scholar] [CrossRef]
- Harrison, P.; Bethel, M.A.; Kennedy, I.; Dinsdale, R.; Coleman, R.; Holman, R.R. Comparison of nine platelet function tests used to determine responses to different aspirin dosages in people with type 2 diabetes. Platelets 2019, 30, 521–529. [Google Scholar] [CrossRef]
- Abaci, A.; Caliskan, M.; Bayram, F.; Yilmaz, Y.; Cetin, M.; Unal, A.; Cetin, S. A new definition of aspirin non-responsiveness by platelet function analyzer-100 and its predictors. Platelets 2006, 17, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Crescente, M.; Di Castelnuovo, A.; Iacoviello, L.; Vermylen, J.; Cerletti, C.; de Gaetano, G. Response variability to aspirin as assessed by the platelet function analyzer (PFA)-100. A systematic review. Thromb. Haemost. 2008, 99, 14–26. [Google Scholar] [CrossRef]
- Mohammedi, K.; Bellili-Muñoz, N.; Marklund, S.L.; Driss, F.; Le Nagard, H.; Patente, T.A.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Marre, M.; et al. Plasma extracellular superoxide dismutase concentration, allelic variations in the SOD3 gene and risk of myocardial infarction and all-cause mortality in people with type 1 and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, M.; Vats, P. Reactive metabolites and antioxidant gene polymorphisms in Type 2 diabetes mellitus. Redox Biol. 2014, 2, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybka, J.; Kupczyk, D.; Kędziora-Kornatowska, K.; Pawluk, H.; Czuczejko, J.; Szewczyk-Golec, K.; Kozakiewicz, M.; Antonioli, M.; Carvalho, L.A.; Kędziora, J. Age-related changes in an antioxidant defense system in elderly patients with essential hypertension compared with healthy controls. Redox Rep. 2011, 16, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, M.; Sharma, T.K.; Singh, I.; Singh, N.; Ghalaut, V.S.; Vardey, S.K.; Shankar, V. Antioxidant enzymes and lipid peroxidation in type 2 diabetes mellitus patients with and without nephropathy. N. Am. J. Med. Sci. 2013, 5, 213–219. [Google Scholar]
- Gawlik, K.; Naskalski, J.W.; Fedak, D.; Pawlica-Gosiewska, D.; Grudzień, U.; Dumnicka, P.; Małecki, M.T.; Solnica, B. Markers of antioxidant defense in patients with type 2 diabetes. Oxid. Med. Cell. Longev. 2016, 2016, 2352361. [Google Scholar] [CrossRef] [Green Version]
- Atli, T.; Keven, K.; Avci, A.; Kutlay, S.; Turkcapar, N.; Varli, M.; Aras, S.; Ertug, E.; Canbolat, O. Oxidative stress and antioxidant status in elderly diabetes mellitus and glucose intolerance patients. Arch. Gerontol. Geriatr. 2004, 39, 269–275. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998, 91, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.C.; Mahoney, C.E.; Coleman Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.-Y.; Tang, S.-S.; Handy, D.E.; Loscalzo, J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 1992, 61, 1175–1212. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, J.; Feng, J. Aspirin resistance mediated by oxidative stress-induced 8-Isoprostaglandin F2. J. Clin. Pharm. Ther. 2019, 44, 823–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davì, G.; Chiarelli, F.; Santilli, F.; Pomilio, M.; Vigneri, S.; Falco, A.; Basili, S.; Ciabattoni, G.; Patrono, C. Enhanced lipid peroxidation and platelet activation in the early phase of type 1 diabetes mellitus: Role of interleukin-6 and disease duration. Circulation 2003, 107, 3199–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarelli, F.; Santilli, F.; Sabatino, G.; Blasetti, A.; Tumini, S.; Cipollone, F.; Mezzetti, A.; Verrotti, A. Effects of vitamin E supplementation on intracellular antioxidant enzyme production in adolescents with type 1 diabetes and early microangiopathy. Pediatr. Res. 2004, 56, 720–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relou, I.A.M.; Hackeng, C.M.; Akkerman, J.-W.N.; Malle, E. Low-density lipoprotein and its effect on human blood platelets. Cell. Mol. Life Sci. 2003, 60, 961–971. [Google Scholar] [CrossRef]
- Barale, C.; Frascaroli, C.; Senkeev, R.; Cavalot, F.; Russo, I. Simvastatin effects on inflammation and platelet activation markers in hypercholesterolemia. Biomed. Res. Int. 2018, 2018, 6508709. [Google Scholar] [CrossRef] [Green Version]
- Carnevale, R.; Bartimoccia, S.; Nocella, C.; Di Santo, S.; Loffredo, L.; Illuminati, G.; Lombardi, E.; Boz, V.; Del Ben, M.; De Marco, L.; et al. LDL oxidation by platelets propagates platelet activation via an oxidative stress-mediated mechanism. Atherosclerosis 2014, 237, 108–116. [Google Scholar] [CrossRef]
- Hovens, M.M.C.; Snoep, J.D.; Groeneveld, Y.; Tamsma, J.T.; Eikenboom, J.C.J.; Huisman, M.V. High levels of low-density lipoprotein cholesterol and triglycerides and suboptimal glycemic control predict diminished ex vivo aspirin responsiveness in patients with Type 2 diabetes. J. Thromb. Haemost. 2007, 5, 1562–1564. [Google Scholar] [CrossRef]
- American Diabetes Association. American Diabetes Association Standards of medical care in diabetes—2014. Diabetes Care 2014, 37 (Suppl. 1), S14–S80. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, C.; Williams, K.; Hunt, K.J.; Haffner, S.M. The National Cholesterol Education Program—Adult Treatment Panel III, International Diabetes Federation, and World Health Organization definitions of the metabolic syndrome as predictors of incident cardiovascular disease and diabetes. Diabetes Care 2007, 30, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Born, G.V. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- Gum, P.A.; Kottke-Marchant, K.; Poggio, E.D.; Gurm, H.; Welsh, P.A.; Brooks, L.; Sapp, S.K.; Topol, E.J. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am. J. Cardiol. 2001, 88, 230–235. [Google Scholar] [CrossRef]
- Harrison, P.; Segal, H.; Blasbery, K.; Furtado, C.; Silver, L.; Rothwell, P.M. Screening for aspirin responsiveness after transient ischemic attack and stroke: Comparison of 2 point-of-care platelet function tests with optical aggregometry. Stroke 2005, 36, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maree, A.O.; Curtin, R.J.; Dooley, M.; Conroy, R.M.; Crean, P.; Cox, D.; Fitzgerald, D.J. Platelet response to low-dose enteric-coated aspirin in patients with stable cardiovascular disease. J. Am. Coll. Cardiol. 2005, 46, 1258–1263. [Google Scholar] [CrossRef] [Green Version]
- Kranzhofer, R.; Ruef, J. Aspirin resistance in coronary artery disease is correlated to elevated markers for oxidative stress but not to the expression of cyclooxygenase (COX) 1/2, a novel COX-1 polymorphism or the PlA(1/2) polymorphism. Platelets 2006, 17, 163–169. [Google Scholar] [CrossRef]
- Basu, S. Isoprostanes: Novel bioactive products of lipid peroxidation. Free Radic. Res. 2004, 38, 105–122. [Google Scholar] [CrossRef]
- Musiek, E.S.; Yin, H.; Milne, G.L.; Morrow, J.D. Recent advances in the biochemistry and clinical relevance of the isoprostane pathway. Lipids 2005, 40, 987–994. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC (n = 61) | T2DM (n = 103) | p | |
|---|---|---|---|
| Female gender, n (%) | 28 (46) | 50 (49) | 0.87 |
| Age (years) | 63.9 ± 7.5 | 64.5 ± 7.0 | 0.58 |
| BMI (kg/m2) | 27.7 ± 3.2 | 29.3 ± 4.0 | 0.008 |
| Fasting Glucose (mg/dl) | 95.9 ± 10.1 | 159.5 ± 42 | 0.0001 |
| HbA1c (%) | 5.9 ± 0.33 | 7.99 ± 1.2 | 0.0001 |
| TC (mg/dl) | 194.2 ± 33.9 | 177.4 ± 37.7 | 0.005 |
| HDL-C (mg/dl) | 52.4 ± 10.4 | 43.8 ± 10.3 | 0.0001 |
| TG (mg/dl) | 126.1 ± 48.5 | 136.3 ± 66.2 | 0.29 |
| LDL-C (mg/dl) | 116.5 ± 32.4 | 103.2 ± 38.7 | 0.019 |
| PLT (x103/µl) | 231 ± 42.3 | 239 ± 48 | 0.3 |
| WBC(x103/µl) | 7.1 ± 1.6 | 7.1 ± 1.8 | 0.92 |
| RBC(x103/µl) | 4.8 ± 0.44 | 4.7 ± 0.50 | 0.5 |
| HCT (%) | 41.9 ± 2.9 | 42.5 ± 3.9 | 0.29 |
| MPV (fl) | 8.4 ± 1.2 | 8.3 ± 0.9 | 0.53 |
| vWF (%) | 130.2 ± 56 | 123 ± 61 | 0.44 |
| Creatinine | 74.6 ± 20.3 | 75.3 ± 27.9 | 0.87 |
| SBP (mmHg) | 129.9 ± 10.6 | 134.3 ± 10.9 | 0.013 |
| DBP (mmHg) | 77.2 ± 7.0 | 76.4 ± 7.3 | 0.48 |
| Previous cerebro-cardiovascular events, n (%) | 28 (43) | 26 (25) | 0.62 |
| Current smokers, n (%) | 14 (23) | 28 (27) | 0.23 |
| Medication use, n (%) | |||
| Statins | 45 (74) | 70 (68) | 0.006 |
| Antihypertensives | 28 (46) | 81 (79) | 0.0001 |
| Insulin | 0 | 24 (23) | N/A |
| Metformin | 0 | 84 (82) | N/A |
| Any oral antidiabetic drugs | 0 | 68 (66) | N/A |
| HC | T2DM | |||||
|---|---|---|---|---|---|---|
| HPR- (n=46) | HPR+ (n=15) | p | HPR- (n=79) | HPR+ (n=24) | p | |
| Female gender, n (%) | 20 (43) | 8 (53) | 0.78 | 40 (51) | 10 (42) | 0.63 |
| Age (years) | 63.9 ± 7.2 | 63.8 ± 8.5 | 0.96 | 65 ± 7.1 | 63 ± 6.7 | 0.21 |
| BMI (kg/m2) | 27.7 ± 3.4 | 27.5 ± 2.6 | 0.79 | 30 ± 4 | 28.5 ± 2.9 | 0.16 |
| Fasting Glucose (mg/dl) | 96.2 ± 11 | 94.9 ± 7.9 | 0.65 | 159 ± 38 | 162.3 ± 54 | 0.81 |
| HbA1c (%) | 5.9 ± 0.4 | 5.8 ± 0.24 | 0.26 | 8 ± 1.1 | 7.9 ± 1.4 | 0.62 |
| TC (mg/dl) | 193 ± 36.3 | 197.5 ± 26.2 | 0.66 | 172 ± 37 | 196 ± 34 | 0.005 |
| HDL-C (mg/dl) | 51.7 ± 10.2 | 54.7 ± 11.2 | 0.32 | 44 ± 11 | 42 ± 9 | 0.27 |
| TG (mg/dl) | 130 ± 51 | 114.1 ± 39.6 | 0.27 | 134 ± 68 | 144 ± 60 | 0.52 |
| LDL-C (mg/dl) | 115.4 ± 35 | 120 ± 23.4 | 0.63 | 99 ± 36 | 120 ± 43 | 0.013 |
| PLT (x103/µl) | 228 ± 42 | 238.3 ± 42.8 | 0.45 | 241 ± 51 | 229 ± 36 | 0.2 |
| WBC(x103/µl) | 7.09 ± 1.4 | 7.2 ± 2.1 | 0.92 | 7.13 ± 1.9 | 6.9 ± 1.6 | 0.58 |
| RBC(x103/µl) | 4.8 ± 0.5 | 4.9 ± 0.41 | 0.48 | 4.7 ± 0.5 | 4.8 ± 0.5 | 0.71 |
| HCT (%) | 41.4 ± 2.4 | 43.4 ± 3.6 | 0.063 | 42 ± 3.7 | 44 ± 4 | 0.17 |
| MPV (fl) | 8.4 ± 1.1 | 8.6 ± 1.4 | 0.51 | 8.3 ± 0.9 | 8.6 ± 1 | 0.22 |
| vWF (%) | 127 ± 59 | 141.6 ± 50 | 0.37 | 122 ± 58 | 126 ± 68 | 0.78 |
| SBP (mmHg) | 130.1 ± 10.6 | 129.3 ± 11 | 0.8 | 135 ± 10 | 133 ± 13 | 0.42 |
| DBP (mmHg) | 77.4 ± 7.1 | 76.7 ± 7 | 0.73 | 77.2 ± 7.4 | 76 ± 6.5 | 0.65 |
| HC (n = 61) | T2DM (n = 103) | p | |
|---|---|---|---|
| SOD (U/mL) | 0.23 ± 0.05 | 0.22 ± 0.05 | 0.4 |
| 8-iso-PGF2α (pmol/mmol creat) | 141 ± 76 | 184 ± 126 | 0.016 |
| sE-Selectin (pg/mL) | 38.3 ± 21 | 48 ± 36 | 0.1 |
| sICAM (ng/mL) | 447 ± 61 | 457 ± 48 | 0.27 |
| sP-Selectin (pg/mL) | 174 ± 96 | 187 ± 62 | 0.298 |
| sCD40L (pg/mL) | 3.4 ± 4.2 | 12 ± 9.3 | 0.0001 |
| IL-6 (pg/mL) | 7.5 ± 4 | 9.7 ± 6 | 0.016 |
| sRAGE (ng/mL) | 1111 ± 640 | 826 ± 327 | 0.0001 |
| TXB2 (ng/mL) | 4.03 ± 2.3 | 4.4 ± 2.9 | 0.381 |
| 11-dhTXB2 (pg/mg creat) | 1388 ± 591 | 1713 ± 1454 | 0.049 |
| HC | T2DM | |||||
|---|---|---|---|---|---|---|
| HPR- | HPR+ | p | HPR- | HPR+ | p | |
| SOD (U/mL) | 0.24 ± 0.04 | 0.18 ± 0.06 | 0.002 | 0.23 ± 0.05 | 0.18 ± 0.04 | 0.0001 |
| 8-iso-PGF2α (pmol/mmol Creat) | 140.1 ± 81 | 142 ± 61 | 0.92 | 161 ± 132 | 259 ± 63 | 0.0001 |
| sE-Selectin (pg/mL) | 37.7 ± 23 | 39.7 ± 13.5 | 0.73 | 46.1 ± 35 | 53.6 ± 37 | 0.39 |
| sICAM (ng/mL) | 450 ± 58 | 440 ± 69.3 | 0.64 | 457.02 ± 52 | 459 ± 36 | 0.89 |
| sP-Selectin (pg/mL) | 155 ± 91 | 245 ± 79.3 | 0.001 | 184 ± 63 | 196 ± 56 | 0.41 |
| sCD40L (pg/mL) | 2.8 ± 2.1 | 5.5 ± 7.7 | 0.052 | 10.8 ± 9.2 | 15.7 ± 9 | 0.003 |
| IL-6 (pg/mL) | 7.5 ± 4.6 | 7.5 ± 3.2 | 0.95 | 9.5 ± 6.1 | 10.2 ± 6.1 | 0.63 |
| sRAGE (ng/mL) | 1225 ± 694 | 807 ± 324 | 0.004 | 841 ± 355 | 777 ± 214 | 0.41 |
| TXB2 (ng/mL) | 3.8 ± 2.3 | 4.85 ± 2.2 | 0.12 | 4.1 ± 2.9 | 5.4 ± 2.6 | 0.59 |
| 11-dhTXB2 (pg/mg creat) | 1270 ± 472 | 1753 ± 770 | 0.037 | 1550 ± 1270 | 2230 ± 1675 | 0.042 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barale, C.; Cavalot, F.; Frascaroli, C.; Bonomo, K.; Morotti, A.; Guerrasio, A.; Russo, I. Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia. Int. J. Mol. Sci. 2020, 21, 4983. https://doi.org/10.3390/ijms21144983
Barale C, Cavalot F, Frascaroli C, Bonomo K, Morotti A, Guerrasio A, Russo I. Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia. International Journal of Molecular Sciences. 2020; 21(14):4983. https://doi.org/10.3390/ijms21144983
Chicago/Turabian StyleBarale, Cristina, Franco Cavalot, Chiara Frascaroli, Katia Bonomo, Alessandro Morotti, Angelo Guerrasio, and Isabella Russo. 2020. "Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia" International Journal of Molecular Sciences 21, no. 14: 4983. https://doi.org/10.3390/ijms21144983
APA StyleBarale, C., Cavalot, F., Frascaroli, C., Bonomo, K., Morotti, A., Guerrasio, A., & Russo, I. (2020). Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia. International Journal of Molecular Sciences, 21(14), 4983. https://doi.org/10.3390/ijms21144983