Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy


Abstract
:1. The Human Flavoproteome
2. Rf Absorption and Cell Delivery
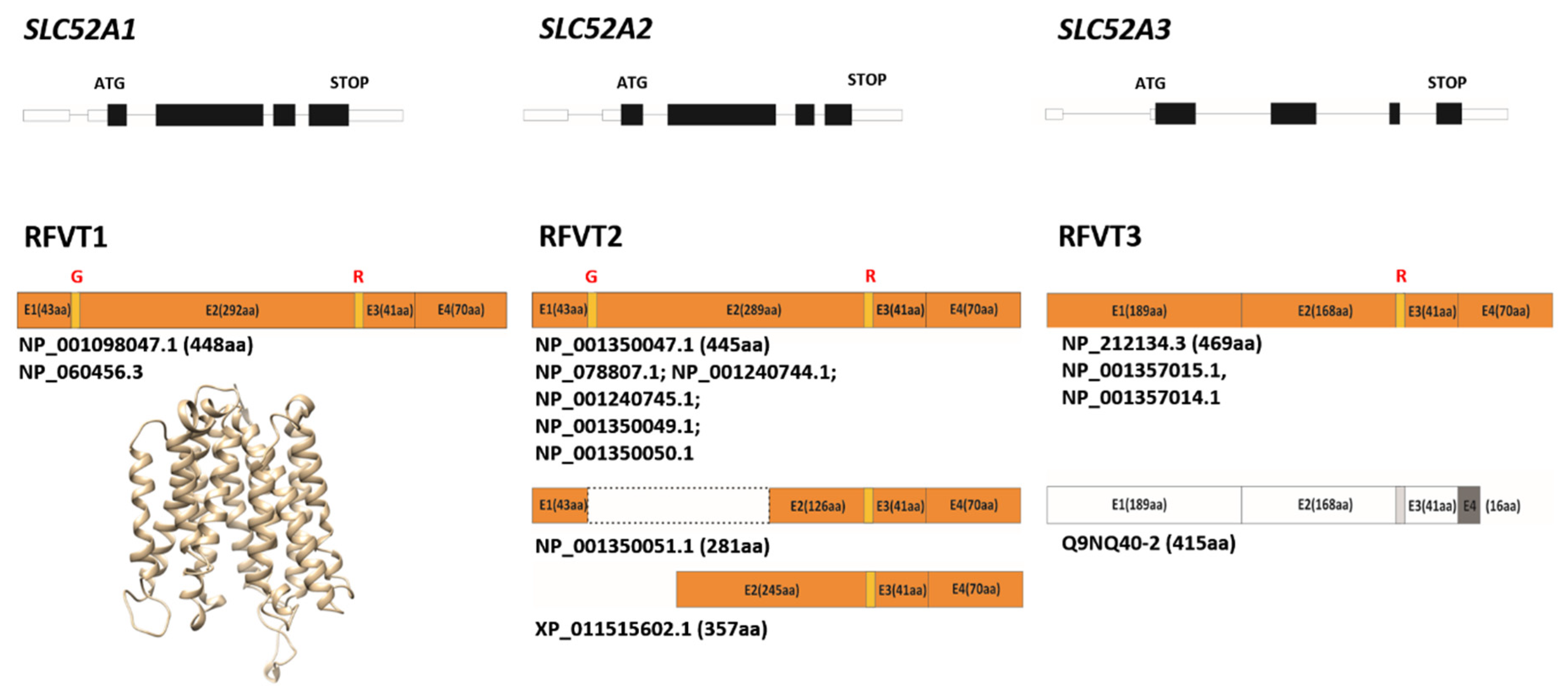
3. Rf Transporters
3.1. Some Molecular Insights on RFVTs
3.2. Rf Transporters: What Else
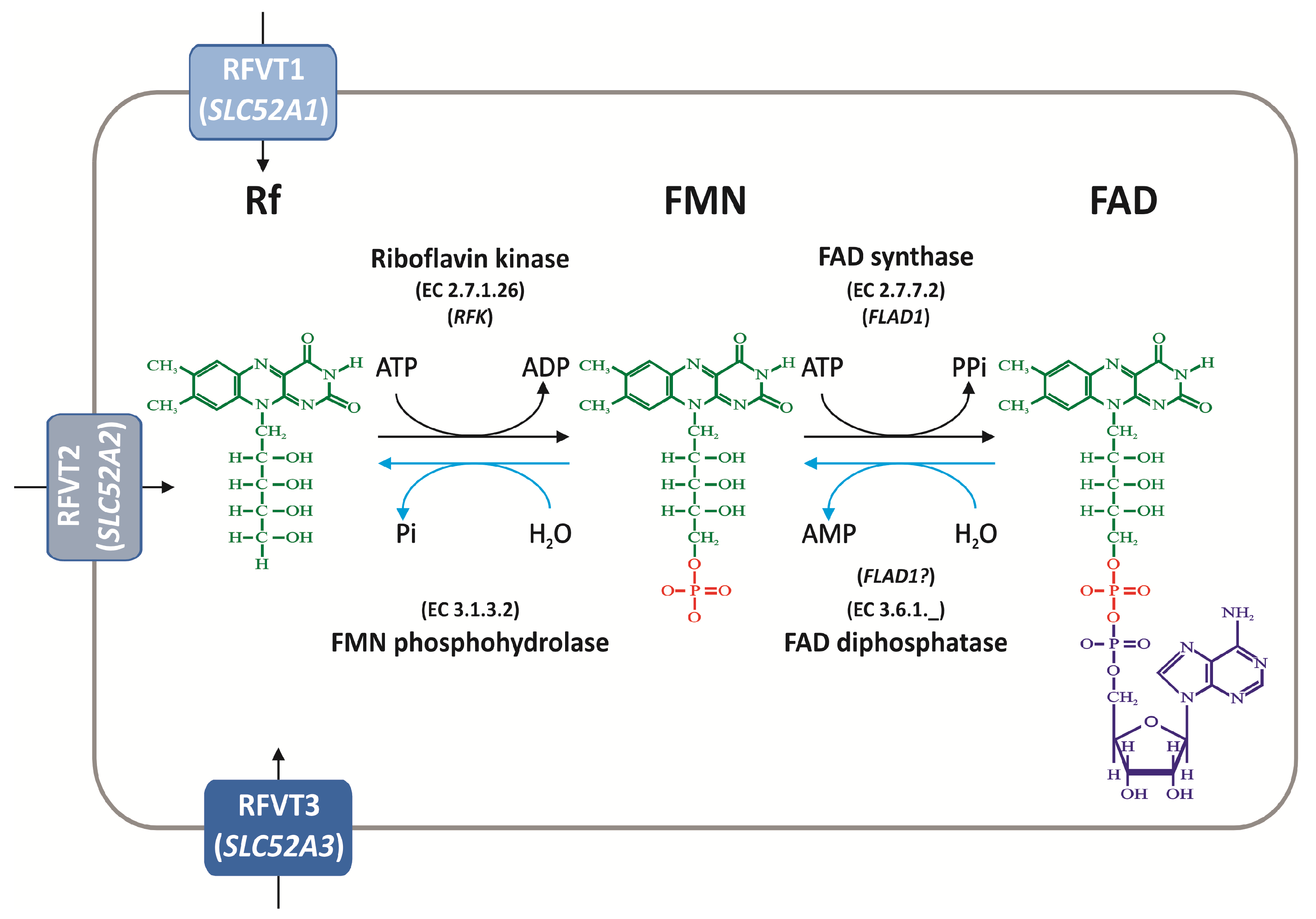
4. Rf Intracellular Homeostasis
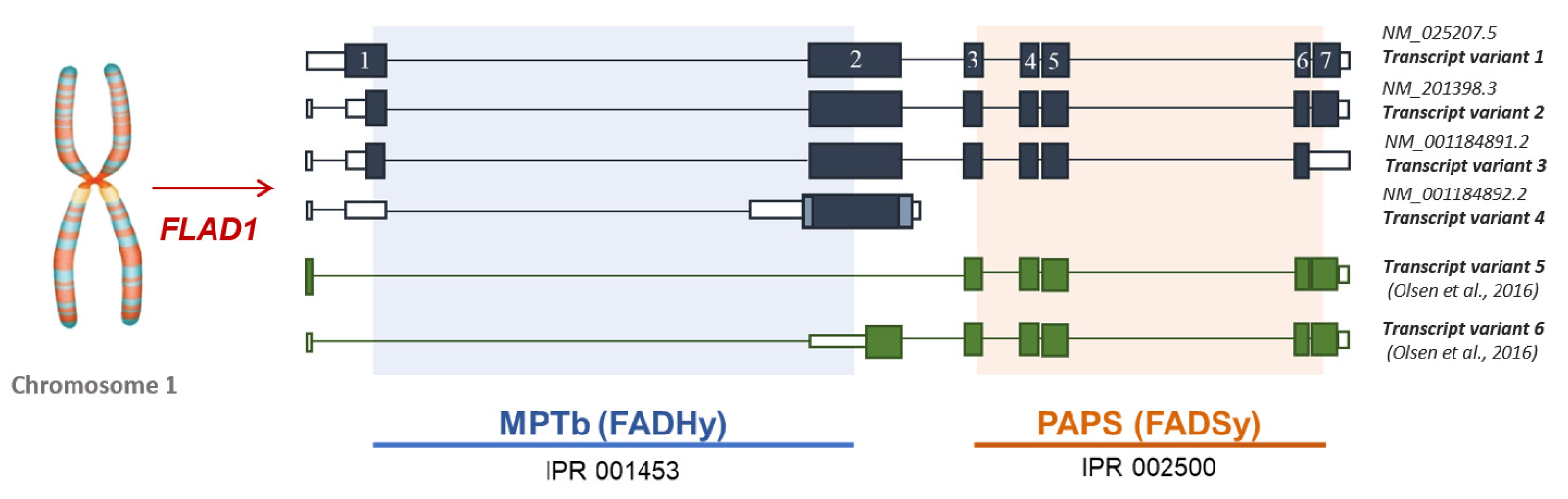
4.1. Biochemical Pathways of FAD Synthesis and Degradation
4.2. The Puzzle of the Splicing Variants of FLAD1 and the Sub-Cellular Origin of FAD
5. Remaining Challenges in Neuronal and Muscular Flavin Homeostasis and their Alterations
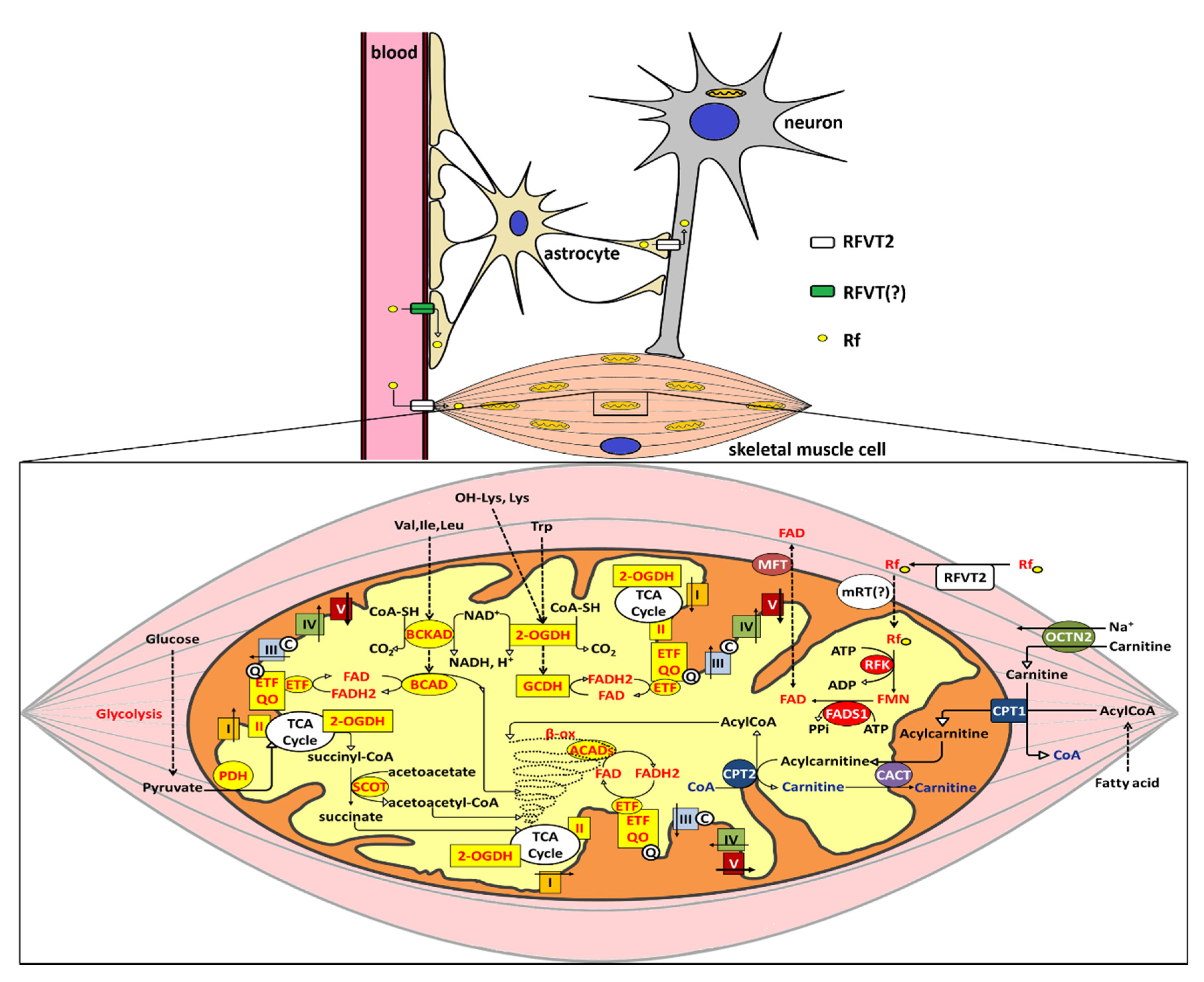
5.1. Rf Neuronal Homeostasis and BVVLS
5.2. Rf Muscular Homeostasis and its Alterations
5.3. A Molecular Rationale for Mitochondrial Flavoproteome Derangement: the Significance of Rf Therapy
6. Model Organisms to Study Flavin Homeostasis Alterations
6.1. Saccharomyces Cerevisiae
6.2. Caenorhabditis Elegans
6.3. Mouse and Drosophila Melanogaster

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Organism | Gene Mutation | Metabolic Dysfunction | References |
|---|---|---|---|
| Caenorhabditis elegans | let-721 | let-721 mutants are maternal effects either lethal or semi-sterile. | [192] |
| Drosophila Melanogaster | Etfdh | Biochemical defects observed in the severe forms of MADD: embryonic accumulation of short-, medium and long-chain acylcarnitines, ETF-QO activity markedly decreased, impaired cofactor association via structural destabilization and consequently enzymatic inactivation. | [193] |
| Zebrafish | Etfdh | Metabolic and mitochondrial dysfunctions, alteration of plasma acylcarnitine and organic acid profiles, reduced oxidative phosphorylation, increased glycolytic flux and the upregulation of the PPARγ-ERK pathway associated to aberrant neural proliferation and motility defects. | [194] |
| Etfa | Pathological and biochemical features similar to those observed for MADD affected individuals, including brain, liver and kidney diseases. An increased signaling of the mechanistic target of rapamycin complex 1 (mTORC1) responsive to treatment with rapamycin was also found. | [195] | |
| Mouse | Etfdh(h)A84T | First RR-MADD mouse model with an Etfdh (h)p.84A > T mutation. The mice, initially normal, developed the clinical and biochemical features typical of MADD under high fat and Rf deficiency diet. Tissues from these mice exhibited a significant decrease of both FAD concentration and ETFDH protein level, which were ameliorated by Rf treatment. | [196] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCG2 | ATP-binding Cassette G2 Transporter |
| AIF | Apoptosis-Inducing Factor |
| AIFM | Apoptosis-Inducing Factor Mitochondria Associated |
| ALS | Amyotrophic Lateral Sclerosis |
| AP-2 | Activating Protein-2 |
| BCRP | Breast Cancer Resistance Protein |
| BVVLS | Brown-Vialetto-Van Laere Syndrome |
| ChIP | Chromatin Immunoprecipitation |
| EGRF | Epidermal Growth Factor Receptor |
| ESCC | Esophageal Squamous Cell Carcinoma |
| ETF | Electron Transfer Flavoprotein |
| ETF-QO | Electron Transfer Flavoprotein-Ubiquinone Oxidoreductase |
| FADS | FAD Synthase |
| FAD | Flavin Adenine Dinucleotide |
| FADDPase | FAD DiPhosphatase |
| FMN | Flavin Mononucleotide |
| LSMFLAD | Flavin Adenine Dinucleotide Synthetase Deficiency |
| KLF | Krupper-Like Factors |
| MADD | Multiple Acyl-CoA Dehydrogenase Deficiency |
| MPTb | MolybdoPterin-Binding |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NF-kB | Nuclear Factor Kappa-light-chain-enhancer of activated B cells |
| Nudix | Nucleoside Diphosphate linked to some other moiety X hydrolase |
| Rf | Riboflavin |
| RFK | Riboflavin Kinase |
| RFVT | Riboflavin Transporter |
| ROS | Reactive Oxygen Species |
| RTD | Riboflavin Transport Deficiency |
| SDH | Succinate Dehydrogenase |
| SLC | Solute Carrier |
| Sp-1 | Stimulating protein-1 |
| TM | Trans-Membrane |
| TNF-α | Tumor Necrosis Factor Alpha |
References
- Lienhart, W.-D.; Gudipati, V.; Macheroux, P. The human flavoproteome. Arch. Biochem. Biophys. 2013, 535, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Piano, V.; Palfey, B.A.; Mattevi, A. Flavins as Covalent Catalysts: New Mechanisms Emerge. Trends Biochem. Sci. 2017, 42, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Heuts, M.P.H.M.; Scrutton, N.S.; McIntire, W.S.; Fraaije, M.W. What’s in a covalent bond? FEBS J. 2009, 276, 3405–3427. [Google Scholar] [CrossRef] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Barile, M.; Giancaspero, T.A.; Brizio, C.; Panebianco, C.; Indiveri, C.; Galluccio, M.; Vergani, L.; Eberini, I.; Gianazza, E. Biosynthesis of flavin cofactors in man: Implications in health and disease. Curr. Pharm. Des. 2013, 19, 2649–2675. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free. Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Edmondson, D.E.; Newton-Vinson, P. The Covalent FAD of Monoamine Oxidase: Structural and Functional Role and Mechanism of the Flavinylation Reaction. Antioxid. Redox Signal. 2001, 3, 789–806. [Google Scholar] [CrossRef]
- Cheng, V.W.T.; Piragasam, R.S.; Rothery, R.; Maklashina, E.; Cecchini, G.; Weiner, J.H. Redox State of Flavin Adenine Dinucleotide Drives Substrate Binding and Product Release in Escherichia coli Succinate Dehydrogenase. Biochemistry 2015, 54, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Decker, K.; Brandsch, R. [39] Determining covalent flavinylation. Methods Enzymol. 1997, 280, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Decker, K.; Brandsch, R. Flavoproteins with a covalent histidyl(N3)-8 alpha-riboflavin linkage. BioFactors 1991, 3, 69–81. [Google Scholar] [PubMed]
- Sevrioukova, I.F. Apoptosis-Inducing Factor: Structure, Function, and Redox Regulation. Antioxid. Redox Signal. 2011, 14, 2545–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragoszewski, P.; Wasilewski, M.; Sakowska, P.; Gornicka, A.; Böttinger, L.; Qiu, J.; Wiedemann, N.; Chacinska, A. Retro-translocation of mitochondrial intermembrane space proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 7713–7718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol–disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, B.P. Biochemical Basis of Oxidative Protein Folding in the Endoplasmic Reticulum. Science 2000, 290, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Joosten, V.; van Berkel, W.J. Flavoenzymes. Curr. Opin. Chem. Biol. 2007, 11, 195–202. [Google Scholar] [CrossRef]
- Hino, S.; Sakamoto, A.; Nagaoka, K.; Anan, K.; Wang, Y.; Mimasu, S.; Umehara, T.; Yokoyama, S.; Kosai, K.-I.; Nakao, M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 2012, 3, 758. [Google Scholar] [CrossRef] [Green Version]
- McCormick, D.B. Two interconnected B vitamins: Riboflavin and pyridoxine. Physiol. Rev. 1989, 69, 1170–1198. [Google Scholar] [CrossRef]
- Powers, H.J. Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [CrossRef]
- Moat, S.J.; Ashfield-Watt, P.A.L.; Powers, H.J.; Newcombe, R.G.; McDowell, I.F.W. Effect of Riboflavin Status on the Homocysteine-lowering Effect of Folate in Relation to the MTHFR (C677T) Genotype. Clin. Chem. 2003, 49, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depeint, F.; Bruce, W.R.; Shangari, N.; Mehta, R.; O’Brien, P.J. Mitochondrial function and toxicity: Role of B vitamins on the one-carbon transfer pathways. Chem. Interact. 2006, 163, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Christodoulou, J.; Rahman, S. Disorders of riboflavin metabolism. J. Inherit. Metab. Dis. 2019, 42, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Ross, N.S.; Hansen, T.P. Riboflavin deficiency is associated with selective preservation of critical flavoenzyme-dependent metabolic pathways. BioFactors 1992, 3, 185–190. [Google Scholar] [PubMed]
- Yonezawa, A.; Inui, K.-I. Novel riboflavin transporter family RFVT/SLC52: Identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol. Asp. Med. 2013, 34, 693–701. [Google Scholar] [CrossRef]
- Yonezawa, A.; Masuda, S.; Katsura, T.; Inui, K.-I. Identification and functional characterization of a novel human and rat riboflavin transporter, RFT1. Am. J. Physiol. Physiol. 2008, 295, C632–C641. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Inoue, K.; Ohta, K.-Y.; Fukatsu, R.; Maeda, J.-Y.; Yoshida, Y.; Yuasa, H. Identification and Functional Characterization of Rat Riboflavin Transporter 2. J. Biochem. 2009, 145, 437–443. [Google Scholar] [CrossRef]
- Yao, Y.; Yonezawa, A.; Yoshimatsu, H.; Masuda, S.; Katsura, T.; Inui, K.-I. Identification and Comparative Functional Characterization of a New Human Riboflavin Transporter hRFT3 Expressed in the Brain. J. Nutr. 2010, 140, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, V.S.; Subramanya, S.B.; Rapp, L.; Marchant, J.S.; Ma, T.Y.; Said, H.M. Differential expression of human riboflavin transporters -1, -2, and -3 in polarized epithelia: A key role for hRFT-2 in intestinal riboflavin uptake. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 3016–3021. [Google Scholar] [CrossRef] [Green Version]
- Karthikeyan, S.; Zhou, Q.; Mseeh, F.; Grishin, N.V.; Osterman, A.L.; Zhang, H. Crystal structure of human riboflavin kinase reveals a beta barrel fold and a novel active site arch. Structure 2003, 11, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Brizio, C.; Galluccio, M.; Wait, R.; Torchetti, E.M.; Bafunno, V.; Accardi, R.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem. Biophys. Res. Commun. 2006, 344, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Yao, Y.; Yonezawa, A.; Imai, S.; Yoshimatsu, H.; Otani, Y.; Omura, T.; Nakagawa, S.; Nakagawa, T.; Matsubara, K. Riboflavin Transporters RFVT/SLC52A Mediate Translocation of Riboflavin, Rather than FMN or FAD, across Plasma Membrane. Biol. Pharm. Bull. 2017, 40, 1990–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, D.B.; Zhang, Z. Cellular Assimilation of Water-Soluble Vitamins in the Mammal: Riboflavin, B6, Biotin, and C. Exp. Biol. Med. 1993, 202, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Galluccio, M.; Miccolis, A.; Leone, P.; Eberini, I.; Iametti, S.; Indiveri, C.; Barile, M. Human FAD synthase is a bi-functional enzyme with a FAD hydrolase activity in the molybdopterin binding domain. Biochem. Biophys. Res. Commun. 2015, 465, 443–449. [Google Scholar] [CrossRef]
- Leone, P.; Galluccio, M.; Brizio, C.; Barbiroli, A.; Iametti, S.; Indiveri, C.; Barile, M. The hidden side of the human FAD synthase 2. Int. J. Biol. Macromol. 2019, 138, 986–995. [Google Scholar] [CrossRef]
- Said, H.M.; Ortiz, A.; Ma, T.Y.; McCloud, E. Riboflavin uptake by the human-derived liver cells Hep G2: Mechanism and regulation. J. Cell. Physiol. 1998, 176, 588–594. [Google Scholar] [CrossRef]
- Ghosal, A.; Said, H.M. Mechanism and regulation of vitamin B2 (riboflavin) uptake by mouse and human pancreatic beta-cells/islets: Physiological and molecular aspects. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G1052–G1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Yonezawa, A.; Yoshimatsu, H.; Omura, T.; Masuda, S.; Matsubara, K. Involvement of riboflavin transporter RFVT2/Slc52a2 in hepatic homeostasis of riboflavin in mice. Eur. J. Pharmacol. 2013, 714, 281–287. [Google Scholar] [CrossRef]
- Innis, W.S.; McCormick, D.B.; Merrill, A.H., Jr. Variations in riboflavin binding by human plasma: Identification of immunoglobulins as the major proteins responsible. Biochem. Med. 1985, 34, 151–165. [Google Scholar] [CrossRef]
- Hustad, S.; McKinley, M.C.; McNulty, H.; Schneede, J.; Strain, J.; Scott, J.M.; Ueland, P.M. Riboflavin, Flavin Mononucleotide, and Flavin Adenine Dinucleotide in Human Plasma and Erythrocytes at Baseline and after Low-Dose Riboflavin Supplementation. Clin. Chem. 2002, 48, 1571–1577. [Google Scholar] [CrossRef]
- Chastain, J.L.; McCormick, D.B. Flavin catabolites: Identification and quantitation in human urine. Am. J. Clin. Nutr. 1987, 46, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Zempleni, J.; Galloway, J.R.; McCormick, D.B. Pharmacokinetics of orally and intravenously administered riboflavin in healthy humans. Am. J. Clin. Nutr. 1996, 63, 54–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, L.; Pang, X.-X.; Lei, F.; Zhang, J.-S.; Wang, W.; Liao, L.-D.; Xu, X.-E.; He, J.-Z.; Wu, J.-Y.; Wu, Z.-Y.; et al. SLC52A3 expression is activated by NF-κB p65/Rel-B and serves as a prognostic biomarker in esophageal cancer. Cell. Mol. Life Sci. 2018, 75, 2643–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Console, L.; Tolomeo, M.; Colella, M.; Barile, M.; Indiveri, C. Reconstitution in Proteoliposomes of the Recombinant Human Riboflavin Transporter 2 (SLC52A2) Overexpressed in E. coli. Int. J. Mol. Sci. 2019, 20, 4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colon-Moran, W.; Argaw, T.; Wilson, C.A. Three cysteine residues of SLC52A1, a receptor for the porcine endogenous retrovirus-A (PERV-A), play a critical role in cell surface expression and infectivity. Virology 2017, 507, 140–150. [Google Scholar] [CrossRef]
- Subramanian, V.S.; Sabui, S.; Teafatiller, T.; Bohl, J.A.; Said, H.M. Structure/functional aspects of the human riboflavin transporter-3 (SLC52A3): Role of the predicted glycosylation and substrate-interacting sites. Am. J. Physiol. Physiol. 2017, 313, C228–C238. [Google Scholar] [CrossRef] [Green Version]
- Foraker, A.B.; Khantwal, C.M.; Swaan, P. Current perspectives on the cellular uptake and trafficking of riboflavin. Adv. Drug Deliv. Rev. 2003, 55, 1467–1483. [Google Scholar] [CrossRef]
- Patel, M.; Vadlapatla, R.K.; Pal, D.; Mandal, A. Molecular and functional characterization of riboflavin specific transport system in rat brain capillary endothelial cells. Brain Res. 2012, 1468, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Said, H.M.; Ortiz, A.; Moyer, M.P.; Yanagawa, N. Riboflavin uptake by human-derived colonic epithelial NCM460 cells. Am. J. Physiol. Physiol. 2000, 278, C270–C276. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, M.; Yamamoto, S.; Murata, T.; Yasujima, T.; Inoue, K.; Ohta, K.-Y.; Yuasa, H. Functional Characteristics of the Human Ortholog of Riboflavin Transporter 2 and Riboflavin-Responsive Expression of Its Rat Ortholog in the Small Intestine Indicate Its Involvement in Riboflavin Absorption. J. Nutr. 2010, 140, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Sabui, S.; Ghosal, A.; Said, H.M. Identification and characterization of 5′-flanking region of the human riboflavin transporter 1 gene (SLC52A1). Gene 2014, 553, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, A.; Sabui, S.; Said, H.M. Identification and characterization of the minimal 5′-regulatory region of the human riboflavin transporter-3 (SLC52A3) in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 2015, 308, C189–C196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhan, R.; Subramanian, V.S.; Said, H.M. Role of MicroRNA-423-5p in posttranscriptional regulation of the intestinal riboflavin transporter-3. Am. J. Physiol. Liver Physiol. 2017, 313, G589–G598. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.S.; Ghosal, A.; Kapadia, R.; Nabokina, S.M.; Said, H.M. Molecular Mechanisms Mediating the Adaptive Regulation of Intestinal Riboflavin Uptake Process. PLOS ONE 2015, 10, e0131698. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, V.S.; Sabui, S.; Heskett, C.W.; Said, H.M. Sodium Butyrate Enhances Intestinal Riboflavin Uptake via Induction of Expression of Riboflavin Transporter-3 (RFVT3). Dig. Dis. Sci. 2018, 64, 84–92. [Google Scholar] [CrossRef]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef]
- Sabui, S.; Subramanian, V.S.; Pham, Q.; Said, H.M. Identification of transmembrane protein 237 as a novel interactor with the intestinal riboflavin transporter-3 (RFVT-3): Role in functionality and cell biology. Am. J. Physiol. Physiol. 2019, 316, C805–C814. [Google Scholar] [CrossRef]
- Anandam, K.Y.; Alwan, O.A.; Subramanian, V.S.; Srinivasan, P.; Kapadia, R.; Said, H.M. Effect of the proinflammatory cytokine TNF-α on intestinal riboflavin uptake: Inhibition mediated via transcriptional mechanism(s). Am. J. Physiol. Physiol. 2018, 315, C653–C663. [Google Scholar] [CrossRef]
- Tutino, V.; DeFrancesco, M.L.; Tolomeo, M.; De Nunzio, V.; Lorusso, D.; Paleni, D.; Caruso, M.G.; Notarnicola, M.; Barile, M. The Expression of Riboflavin Transporters in Human Colorectal Cancer. Anticancer Res. 2018, 38, 2659–2667. [Google Scholar] [CrossRef]
- Pinto, J.T.; Zempleni, J. Riboflavin. Adv. Nutr. 2016, 7, 973–975. [Google Scholar] [CrossRef] [Green Version]
- Horsey, A.J.; Cox, M.H.; Sarwat, S.; Kerr, I.D. The multidrug transporter ABCG2: Still more questions than answers. Biochem. Soc. Trans. 2016, 44, 824–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaming, M.L.; Lagas, J.S.; Schinkel, A.H. Physiological and pharmacological roles of ABCG2 (BCRP): Recent findings in Abcg2 knockout mice. Adv. Drug Deliv. Rev. 2009, 61, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Brizio, C.; Valenti, D.; De Virgilio, C.; Passarella, S. The riboflavin/FAD cycle in rat liver mitochondria. JBIC J. Biol. Inorg. Chem. 2000, 267, 4888–4900. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Locato, V.; De Pinto, M.C.; De Gara, L.; Barile, M. The occurrence of riboflavin kinase and FAD synthetase ensures FAD synthesis in tobacco mitochondria and maintenance of cellular redox status. FEBS J. 2008, 276, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Merrill, A.H.; McCormick, D.B. Probable reaction mechanisms of flavokinase and FAD synthetase from rat liver. Arch. Biochem. Biophys. 1990, 278, 125–130. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Zhou, Q.; Osterman, A.L.; Zhang, H. Ligand Binding-Induced Conformational Changes in Riboflavin Kinase: Structural Basis for the Ordered Mechanism. Biochemistry 2003, 42, 12532–12538. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-S.; McCormick, D.B. Thyroid hormone regulation of flavocoenzyme biosynthesis. Arch. Biochem. Biophys. 1985, 237, 197–201. [Google Scholar] [CrossRef]
- Yazdanpanah, B.; Wiegmann, K.; Tchikov, V.; Krut, O.; Pongratz, C.; Schramm, M.; Kleinridders, A.; Wunderlich, T.; Kashkar, H.; Utermöhlen, O.; et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 2009, 460, 1159–1163. [Google Scholar] [CrossRef]
- Schramm, M.; Wiegmann, K.; Schramm, S.; Gluschko, A.; Herb, M.; Utermöhlen, O.; Krönke, M. Riboflavin (vitamin B2) deficiency impairs NADPH oxidase 2 (Nox2) priming and defense againstListeria monocytogenes. Eur. J. Immunol. 2013, 44, 728–741. [Google Scholar] [CrossRef]
- Hirano, A.; Braas, D.; Fu, Y.-H.; Ptáček, L.J. FAD Regulates CRYPTOCHROME Protein Stability and Circadian Clock in Mice. Cell Rep. 2017, 19, 255–266. [Google Scholar] [CrossRef]
- Galluccio, M.; Brizio, C.; Torchetti, E.M.; Ferranti, P.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli, purification and characterization of isoform 2 of human FAD synthetase. Protein Expr. Purif. 2007, 52, 175–181. [Google Scholar] [CrossRef]
- Torchetti, E.M.; Brizio, C.; Colella, M.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Roberti, M.; Barile, M. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion 2010, 10, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Galluccio, M.; Barbiroli, A.; Eberini, I.; Tolomeo, M.; Vrenna, F.; Gianazza, E.; Iametti, S.; Bonomi, F.; Indiveri, C.; et al. Bacterial Production, Characterization and Protein Modeling of a Novel Monofuctional Isoform of FAD Synthase in Humans: An Emergency Protein? Molecules 2018, 23, 116. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Galluccio, M.; Quarta, S.; Anoz-Carbonell, E.; Medina, M.; Indiveri, C.; Barile, M. Mutation of Aspartate 238 in FAD Synthase Isoform 6 Increases the Specific Activity by Weakening the FAD Binding. Int. J. Mol. Sci. 2019, 20, 6203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miccolis, A.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Barile, M. Bacterial Over-Expression and Purification of the 3′phosphoadenosine 5′phosphosulfate (PAPS) Reductase Domain of Human FAD Synthase: Functional Characterization and Homology Modeling. Int. J. Mol. Sci. 2012, 13, 16880–16898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miccolis, A.; Galluccio, M.; Nitride, C.; Giancaspero, T.A.; Ferranti, P.; Iametti, S.; Indiveri, C.; Bonomi, F.; Barile, M. Significance of redox-active cysteines in human FAD synthase isoform 2. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2014, 1844, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.H.; Sa, N.; Saeheng, S.; Raffaelli, N.; Roje, S. Characterization of a non-nudix pyrophosphatase points to interplay between flavin and NAD(H) homeostasis in Saccharomyces cerevisiae. PLOS ONE 2018, 13, e0198787. [Google Scholar] [CrossRef] [Green Version]
- Giancaspero, T.A.; Colella, M.; Brizio, C.; Difonzo, G.; Fiorino, G.M.; Leone, P.; Brandsch, R.; Bonomi, F.; Iametti, S.; Barile, M. Remaining challenges in cellular flavin cofactor homeostasis and flavoprotein biogenesis. Front. Chem. 2015, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Torchetti, E.M.; Bonomi, F.; Galluccio, M.; Gianazza, E.; Giancaspero, T.A.; Iametti, S.; Indiveri, C.; Barile, M. Human FAD synthase (isoform 2): A component of the machinery that delivers FAD to apo-flavoproteins. FEBS J. 2011, 278, 4434–4449. [Google Scholar] [CrossRef]
- Olsen, R.; Konarikova, E.; Giancaspero, T.A.; Mosegaard, S.; Boczonadi, V.; Matakovic, L.; Veauville-Merllié, A.; Terrile, C.; Schwarzmayr, T.; Haack, T.B.; et al. Riboflavin-Responsive and -Non-responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2016, 98, 1130–1145. [Google Scholar] [CrossRef] [Green Version]
- Giancaspero, T.A.; Busco, G.; Panebianco, C.; Carmone, C.; Miccolis, A.; Liuzzi, G.M.; Colella, M.; Barile, M. FAD Synthesis and Degradation in the Nucleus Create a Local Flavin Cofactor Pool*. J. Biol. Chem. 2013, 288, 29069–29080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, M.; Bannister, A.J. Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol. Metab. 2020, 38, 100942. [Google Scholar] [CrossRef] [PubMed]
- Barile, M.; Brizio, C.; Virgilio, C.; Delfine, S.; Quagliariello, E.; Passarella, S. Flavin Adenine Dinucleotide and Flavin Mononucleotide Metabolism in Rat Liver. The Occurrence of FAD Pyrophosphatase and FMN Phosphohydrolase in Isolated Mitochondria. JBIC J. Biol. Inorg. Chem. 1997, 249, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Vergani, L.; Barile, M.; Angelini, C.; Burlina, A.B.; Nijtmans, L.; Freda, M.P.; Brizio, C.; Zerbetto, E.; Dabbeni-Sala, F. Riboflavin therapy. Biochemical heterogeneity in two adult lipid storage myopathies. Brain 1999, 122, 122. [Google Scholar]
- Titus, S.A.; Moran, R.G. Retrovirally mediated complementation of the glyB phenotype. Cloning of a human gene encoding the carrier for entry of folates into mitochondria. J. Biol. Chem. 2000, 275, 36811–36817. [Google Scholar] [CrossRef] [Green Version]
- Tzagoloff, A.; Jang, J.; Glerum, D.M.; Wu, M. FLX1Codes for a Carrier Protein Involved in Maintaining a Proper Balance of Flavin Nucleotides in Yeast Mitochondria. J. Biol. Chem. 1996, 271, 7392–7397. [Google Scholar] [CrossRef] [Green Version]
- Spaan, A.N.; Ijlst, L.; Van Roermund, C.W.; Wijburg, F.A.; Wanders, R.J.; Waterham, H.R. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2005, 86, 441–447. [Google Scholar] [CrossRef]
- Bafunno, V.; Giancaspero, T.A.; Brizio, C.; Bufano, D.; Passarella, S.; Boles, E.; Barile, M. Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: Involvement of the Flx1p carrier in FAD export. J. Biol. Chem. 2004, 279, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Schiff, M.; Veauville-Merllié, A.; Su, C.H.; Tzagoloff, A.; Rak, M.; De Baulny, H.O.; Boutron, A.; Smedts-Walters, H.; Romero, N.B.; Rigal, O.; et al. SLC25A32 Mutations and Riboflavin-Responsive Exercise Intolerance. N. Engl. J. Med. 2016, 374, 795–797. [Google Scholar] [CrossRef] [Green Version]
- Hellebrekers, D.M.; Sallevelt, S.C.E.H.; Theunissen, T.E.J.; Hendrickx, A.T.M.; Gottschalk, R.W.; Hoeijmakers, J.G.J.; Habets, D.D.; Bierau, J.; Schoonderwoerd, K.G.; Smeets, H.J.M. Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur. J. Hum. Genet. 2017, 25, 886–888. [Google Scholar] [CrossRef]
- Santoro, V.; Kovalenko, I.; Vriens, K.; Christen, S.; Bernthaler, A.; Haegebarth, A.; Fendt, S.-M.; Christian, S. SLC25A32 sustains cancer cell proliferation by regulating flavin adenine nucleotide (FAD) metabolism. Oncotarget 2020, 11, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, R. Vitamin Transport Diseases of Brain: Focus on Folates, Thiamine and Riboflavin. Brain Disord. Ther. 2014, 3. [Google Scholar] [CrossRef] [Green Version]
- Ciccolella, M.; Corti, S.; Catteruccia, M.; Petrini, S.; Tozzi, G.; Rizza, T.; Carrozzo, R.; Nizzardo, M.; Bordoni, A.; Ronchi, D.; et al. Riboflavin transporter 3 involvement in infantile Brown-Vialetto-Van Laere disease: Two novel mutations. J. Med. Genet. 2012, 50, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udhayabanu, T.; Manole, A.; Rajeshwari, M.; Varalakshmi, P.; Houlden, H.; AshokKumar, B. Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Clin. Med. 2017, 6, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, R. Riboflavin Homeostasis in the Central Nervous System. J. Neurochem. 1980, 35, 202–209. [Google Scholar] [CrossRef]
- Spector, R. Riboflavin transport in the central nervous system. Characterization and effects of drugs. J. Clin. Investig. 1980, 66, 821–831. [Google Scholar] [CrossRef]
- Freeman, M.R. Specification and Morphogenesis of Astrocytes. Science 2010, 330, 774–778. [Google Scholar] [CrossRef] [Green Version]
- Green, P.; Wiseman, M.; Crow, Y.J.; Houlden, H.; Riphagen, S.; Lin, J.-P.; Raymond, F.L.; Childs, A.-M.; Sheridan, E.; Edwards, S.; et al. Brown-Vialetto-Van Laere Syndrome, a Ponto-Bulbar Palsy with Deafness, Is Caused by Mutations in C20orf54. Am. J. Hum. Genet. 2010, 86, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Dipti, S.; Childs, A.-M.; Livingston, J.H.; Aggarwal, A.; Miller, M.; Williams, C.; Crow, Y.J. Brown–Vialetto–Van Laere syndrome; variability in age at onset and disease progression highlighting the phenotypic overlap with Fazio-Londe disease. Brain Dev. 2005, 27, 443–446. [Google Scholar] [CrossRef]
- Haack, T.B.; Makowski, C.; Yao, Y.; Graf, E.; Hempel, M.; Wieland, T.; Tauer, U.; Ahting, U.; Mayr, J.A.; Freisinger, P.; et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012, 35, 943–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.O.; Gibbs, J.R.; Megarbane, A.; Urtizberea, J.A.; Hernandez, D.G.; Foley, A.R.; Arepalli, S.; Pandraud, A.; Simon-Sanchez, J.; Clayton, P.; et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012, 135, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, B.; Bosch, A.M.; Houlden, H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J. Inherit. Metab. Dis. 2019, 42, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Amir, F.; Atzinger, C.; Massey, K.; Greinwald, J.; Hunter, L.L.; Ulm, E.; Kettler, M. The Clinical Journey of Patients with Riboflavin Transporter Deficiency Type 2. J. Child. Neurol. 2019, 35, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Abeling, N.G.G.M.; Ijlst, L.; Knoester, H.; Van Der Pol, W.L.; Stroomer, A.E.M.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2010, 34, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Fogel, B.L. Successful treatment of a genetic childhood ataxia due to riboflavin transporter deficiency. Cerebellum Ataxias 2018, 5, 12. [Google Scholar] [CrossRef]
- Bamaga, A.K.; Maamari, R.N.; Culican, S.M.; Shinawi, M.; Golumbek, P.T. Child Neurology: Brown-Vialetto-Van Laere syndrome: Dramatic visual recovery after delayed riboflavin therapy. Neurology 2018, 91, 938–941. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Shi, Z.; Yan, H.; Wang, X.-D.; Yang, Y.; Xiong, H.; Gu, Q.; Wu, Y.; Jiang, Y.; Wang, J. A Chinese pedigree with Brown-Vialetto-Van Laere syndrome due to two novel mutations of SLC52A2 gene: Clinical course and response to riboflavin. BMC Med. Genet. 2019, 20, 76. [Google Scholar] [CrossRef]
- Gorcenco, S.; Vaz, F.M.; Tracewska-Siemiatkowska, A.; Tranebjærg, L.; Cremers, F.P.; Ygland, E.; Kicsi, J.; Rendtorff, N.D.; Möller, C.; Kjellström, U.; et al. Oral therapy for riboflavin transporter deficiency - What is the regimen of choice? Park. Relat. Disord. 2019, 61, 245–247. [Google Scholar] [CrossRef]
- Abbas, Q.; Jafri, S.K.; Ishaque, S.; Rahman, A.J. Brown-Vialetto-Van Laere syndrome: A novel diagnosis to a common presentation. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Garg, M.; Kulkarni, S.D.; Hegde, A.U.; Shah, K.N. Riboflavin treatment in genetically proven Brown–Vialetto–Van Laere syndrome. J. Pediatr. Neurosci. 2018, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, B.; Topcu, M.T.; Ciprut, A. A Case with Brown-Vialetto-Van Laere Syndrome: A Sudden Onset Auditory Neuropathy Spectrum Disorder. Turk. Arch. Otorhinolaryngol. 2019, 57, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, B.; Bakhshandeh, M.K.; Navaeifar, M.R.; Abbaskhanian, A.; Soveizi, M.; Geravandpoor, S.; Mahdieh, N. Brown-Vialetto-Van Laere syndrome and Fazio-Londe syndrome: A novel mutation and in silico analyses. J. Clin. Neurosci. 2020, 72, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Carreau, C.; Lenglet, T.; Mosnier, I.; Lahlou, G.; Fargeot, G.; Weiss, N.; Demeret, S.; Salachas, F.; Veauville-Merllié, A.; Acquaviva, C.; et al. A juvenile ALS-like phenotype dramatically improved after high-dose riboflavin treatment. Ann. Clin. Transl. Neurol. 2020, 7, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2013, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Golpich, M.; Amini, E.; Mohamed, Z.; Ali, R.A.; Ibrahim, N.M.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2016, 23, 5–22. [Google Scholar] [CrossRef]
- Manole, A.; Jaunmuktane, Z.; Hargreaves, I.; Ludtmann, M.H.R.; Salpietro, V.; Bello, O.; Pope, S.A.; Pandraud, A.; Horga, A.; Scalco, R.; et al. Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain 2017, 140, 2820–2837. [Google Scholar] [CrossRef] [Green Version]
- Foley, A.R.; Menezes, M.P.; Pandraud, A.; Gonzalez, M.A.; Al-Odaib, A.; Abrams, A.J.; Sugano, K.; Yonezawa, A.; Manzur, A.Y.; Burns, J.; et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 2013, 137, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Chaya, S.; Zampoli, M.; Gray, D.; Booth, J.; Riordan, G.; Ndondo, A.; Fieggen, K.; Rusch, J.; Van Der Watt, G.; Pillay, K.; et al. The First Case of Riboflavin Transporter Deficiency in sub-Saharan Africa. Semin. Pediatr. Neurol. 2018, 26, 10–14. [Google Scholar] [CrossRef]
- Nimmo, G.A.M.; Ejaz, R.; Cordeiro, D.; Kannu, P.; Mercimek-Andrews, S. Riboflavin transporter deficiency mimicking mitochondrial myopathy caused by complex II deficiency. Am. J. Med. Genet. Part. A 2017, 176, 399–403. [Google Scholar] [CrossRef]
- Rizzo, F.; Ramirez, A.; Compagnucci, C.; Salani, S.; Melzi, V.; Bordoni, A.; Fortunato, F.; Niceforo, A.; Bresolin, N.; Comi, G.P.; et al. Genome-wide RNA-seq of iPSC-derived motor neurons indicates selective cytoskeletal perturbation in Brown–Vialetto disease that is partially rescued by riboflavin. Sci. Rep. 2017, 7, 46271. [Google Scholar] [CrossRef] [PubMed]
- Difonzo, G.; Giancaspero, T.A.; Busco, G.; Panebianco, C.; Colella, M.; Barile, M. A Novel Subcellular Localization of FAD Synthase in Neuronal Cells Models and Possible Consequences of Altered Flavin Homeostasis. Unpublished work. 2015. [Google Scholar]
- Lin, J.; Diamanduros, A.; Chowdhury, S.A.; Scelsa, S.; Latov, N.; Sadiq, S.A. Specific electron transport chain abnormalities in amyotrophic lateral sclerosis. J. Neurol. 2009, 256, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, V.C.; Giancaspero, T.A.; Gianazza, E.; Banfi, C.; Barile, M.; De Giorgi, C. Silencing of FAD synthase gene in Caenorhabditis elegans upsets protein homeostasis and impacts on complex behavioral patterns. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Vergani, L.; Wait, R.; Brizio, C.; Brambilla, D.; Begum, S.; Giancaspero, T.A.; Conserva, F.; Eberini, I.; Bufano, D.; et al. Coordinated and reversible reduction of enzymes involved in terminal oxidative metabolism in skeletal muscle mitochondria from a riboflavin-responsive, multiple acyl-CoA dehydrogenase deficiency patient. Electrophoresis 2006, 27, 1182–1198. [Google Scholar] [CrossRef]
- Rhead, W.; Roettger, V.; Marshali, T.; Amendt, B. Multiple Acyl-Coenzyme A Dehydrogenation Disorder Responsive to Riboflavin: Substrate Oxidation, Flavin Metabolism, and Flavoenzyme Activities in Fibroblasts. Pediatr. Res. 1993, 33, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryder, B.; Tolomeo, M.; Nochi, Z.; Colella, M.; Barile, M.; Olsen, R.; Inbar-Feigenberg, M. A Novel Truncating FLAD1 Variant, Causing Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) in an 8-Year-Old Boy. JIMD Rep. 2018, 45, 37–44. [Google Scholar] [CrossRef]
- Taylor, R.W.; Pyle, A.; Griffin, H.R.; Blakely, E.L.; Duff, J.; He, L.; Smertenko, T.; Alston, C.L.; Neeve, V.C.; Best, A.; et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014, 312, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Yıldız, Y.; Olsen, R.K.J.; Sivri, H.S.; Akçören, Z.; Nygaard, H.H.; Tokatlı, A.; Yıldız, Y. Post-mortem detection of FLAD1 mutations in 2 Turkish siblings with hypotonia in early infancy. Neuromuscul. Disord. 2018, 28, 787–790. [Google Scholar] [CrossRef]
- Mosegaard, S.; DiPace, G.; Bross, P.; Carlsen, J.; Gregersen, N.; Olsen, R.K. Riboflavin Deficiency—Implications for General Human Health and Inborn Errors of Metabolism. Int. J. Mol. Sci. 2020, 21, 3847. [Google Scholar] [CrossRef]
- Panebianco, C.; Barile, M. Expression and subcellular localization of FAD synthase. Relationships with Human Pathologies. Unpublished work. 2013. [Google Scholar]
- Zerbetto, E.; Vergani, L.; Dabbeni-Sala, F. Quantification of muscle mitochondrial oxidative phosphorylation enzymes via histochemical staining of blue native polyacrylamide gels. Electrophoresis 1997, 18, 2059–2064. [Google Scholar] [CrossRef]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2005, 63, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Sim, K.; Carpenter, K.; Rhead, W.; Ho, G.; Olsen, R.; Christodoulou, J. Transient multiple acyl-CoA dehydrogenation deficiency in a newborn female caused by maternal riboflavin deficiency. Mol. Genet. Metab. 2007, 92, 109–114. [Google Scholar] [CrossRef]
- Ho, G.; Yonezawa, A.; Masuda, S.; Inui, K.-I.; Sim, K.G.; Carpenter, K.; Olsen, R.; Mitchell, J.; Rhead, W.J.; Peters, G.; et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat. 2010, 32, E1976–E1984. [Google Scholar] [CrossRef] [PubMed]
- Mosegaard, S.; Bruun, G.H.; Flyvbjerg, K.F.; Bliksrud, Y.T.; Gregersen, N.; Dembic, M.; Annexstad, E.; Tangeraas, T.; Olsen, R.; Andresen, B.S. An intronic variation in SLC52A1 causes exon skipping and transient riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Mol. Genet. Metab. 2017, 122, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Andresen, B.S.; Pedersen, C.B.; Olsen, R.; Corydon, T.J.; Bross, P. Mitochondrial fatty acid oxidation defects—remaining challenges. J. Inherit. Metab. Dis. 2008, 31, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Kompare, M.; Rizzo, W.B. Mitochondrial Fatty-Acid Oxidation Disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. [Google Scholar] [CrossRef]
- Ames, B.N.; Elson-Schwab, I.; Silver, E.A. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): Relevance to genetic disease and polymorphisms. Am. J. Clin. Nutr. 2002, 75, 616–658. [Google Scholar] [CrossRef] [Green Version]
- Henriques, B.; Rodrigues, J.V.; Olsen, R.; Bross, P.; Gomes, C.M. Role of Flavinylation in a Mild Variant of Multiple Acyl-CoA Dehydrogenation Deficiency. J. Biol. Chem. 2008, 284, 4222–4229. [Google Scholar] [CrossRef] [Green Version]
- Lucas, T.G.; Henriques, B.J.; Gomes, C.M. Conformational analysis of the riboflavin-responsive ETF:QO-p.Pro456Leu variant associated with mild multiple acyl-CoA dehydrogenase deficiency. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140393. [Google Scholar] [CrossRef]
- Lin, Y.-F.; Haynes, C.M. Metabolism and the UPR(mt). Mol. Cell 2016, 61, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, N.; Frerman, F.E.; Corydon, T.J.; Palmfeldt, J.; Bross, P.; Gregersen, N.; Olsen, R. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2012, 21, 3435–3448. [Google Scholar] [CrossRef] [Green Version]
- Giancaspero, T.A.; Wait, R.; Boles, E.; Barile, M. Succinate dehydrogenase flavoprotein subunit expression in Saccharomyces cerevisiae- involvement of the mitochondrial FAD transporter, Flx1p. FEBS J. 2008, 275, 1103–1117. [Google Scholar] [CrossRef]
- Troulinaki, K.; Büttner, S.; Marsal Cots, A.; Maida, S.; Meyer, K.; Bertan, K.; Gioran, A.; Piazzesi, A.; Fornarelli, A.; Nicotera, P.; et al. WAH–1/AIF regulates mitochondrial oxidative phosphorylation in the nematode Caenorhabditis elegans. Cell Death Discov. 2018, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Hangen, E.; Féraud, O.; Lachkar, S.; Mou, H.; Doti, N.; Fimia, G.M.; Lam, N.-V.; Zhu, C.; Godin, I.; Müller, K.; et al. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol. Cell 2015, 58, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.; Buettner, S.; Ghezzi, D.; Zeviani, M.; Bano, D.; Nicotera, P. Loss of apoptosis-inducing factor critically affects MIA40 function. Cell Death Dis. 2015, 6, e1814. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, L.; Calogero, A.M.; Pandini, V.; Vanoni, M.A.; Sevrioukova, I.F.; Aliverti, A. Key Role of the Adenylate Moiety and Integrity of the Adenylate-Binding Site for the NAD+/H Binding to Mitochondrial Apoptosis-Inducing Factor. Biochemistry 2015, 54, 6996–7009. [Google Scholar] [CrossRef]
- Sorrentino, L.; Cossu, F.; Milani, M.; Aliverti, A.; Mastrangelo, E. Structural bases of the altered catalytic properties of a pathogenic variant of apoptosis inducing factor. Biochem. Biophys. Res. Commun. 2017, 490, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, R.; Ferreira, P.; Marcuello, C.; Usón, A.; Miramar, M.D.; Peleato, M.L.; Lostao, A.; Susin, S.A.; Medina, M.; Medina, M. Key Residues Regulating the Reductase Activity of the Human Mitochondrial Apoptosis Inducing Factor. Biochemistry 2015, 54, 5175–5184. [Google Scholar] [CrossRef]
- Ghezzi, D.; Sevrioukova, I.; Invernizzi, F.; Lamperti, C.; Mora, M.; D’Adamo, A.P.; Novara, F.; Zuffardi, O.; Uziel, G.; Zeviani, M. Severe X-Linked Mitochondrial Encephalomyopathy Associated with a Mutation in Apoptosis-Inducing Factor. Am. J. Hum. Genet. 2010, 86, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Sevrioukova, I.F. Structure/Function Relations in AIFM1 Variants Associated with Neurodegenerative Disorders. J. Mol. Biol. 2016, 428, 3650–3665. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Guan, J.; Ealy, M.; Zhang, Q.; Wang, D.; Wang, H.; Zhao, Y.; Shen, Z.; Campbell, C.; Wang, F.; et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. J. Med. Genet. 2015, 52, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Kettwig, M.; Schubach, M.; Zimmermann, F.A.; Klinge, L.; Mayr, J.A.; Biskup, S.; Sperl, W.; Gärtner, J.; Huppke, D.P. From ventriculomegaly to severe muscular atrophy: Expansion of the clinical spectrum related to mutations in AIFM1. Mitochondrion 2015, 21, 12–18. [Google Scholar] [CrossRef]
- Gregersen, N.; Kølvraa, S.; Mortensen, P.B.; Rasmussen, K. C6-C10-dicarboxylic aciduria: Biochemical considerations in relation to diagnosis of beta-oxidation defects. Scand. J. Clin. Lab. Investig. Suppl. 1982, 161, 15–27. [Google Scholar]
- Antozzi, C.; Garavaglia, B.; Mora, M.; Rimoldi, M.; Morandi, L.; Ursino, E.; DiDonato, S. Late-onset riboflavin-responsive myopathy with combined multiple acyl coenzyme A dehydrogenase and respiratory chain deficiency. Neurology 1994, 44, 2153. [Google Scholar] [CrossRef]
- Olsen, R.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2010, 1797, 1910–1916. [Google Scholar] [CrossRef]
- Cornelius, N.; Byron, C.; Hargreaves, I.; Fernandez-Guerra, P.; Furdek, A.K.; Land, J.; Radford, W.W.; Frerman, F.; Corydon, T.J.; Gregersen, N.; et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2013, 22, 3819–3827. [Google Scholar] [CrossRef]
- Rocha, H.; Ferreira, R.; Carvalho, J.; Vitorino, R.; Santa, C.; Lopes, L.; Gregersen, N.; Vilarinho, L.; Amado, F. Characterization of mitochondrial proteome in a severe case of ETF-QO deficiency. J. Proteom. 2011, 75, 221–228. [Google Scholar] [CrossRef]
- Van Rijt, W.J.; Ferdinandusse, S.; Giannopoulos, P.; Ruiter, J.P.N.; De Boer, L.; Bosch, A.M.; Huidekoper, H.H.; Rubio-Gozalbo, M.E.; Visser, G.; Williams, M.; et al. Prediction of disease severity in multiple acyl-CoA dehydrogenase deficiency: A retrospective and laboratory cohort study. J. Inherit. Metab. Dis. 2019, 42, 878–889. [Google Scholar] [CrossRef]
- Gudipati, V.; Koch, K.; Lienhart, W.-D.; Macheroux, P. The flavoproteome of the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA) Bioenergy 2013, 1844, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Perl, M.; Kearney, E.B.; Singer, T.P. Transport of riboflavin into yeast cells. J. Biol. Chem. 1976, 251, 3221–3228. [Google Scholar] [PubMed]
- Sibirnyĭ, A.A.; Shavlovskiĭ, G.M.; Ksheminskaia, G.P.; Orlovskaia, A.G. [Active transport of riboflavin in the yeast Pichia guilliermondii. Detection and some properties of the cryptic riboflavin permease]. Биохимия 1977, 42, 1841–1851. [Google Scholar]
- Schwechheimer, S.K.; Park, E.Y.; Revuelta, J.L.; Becker, J.; Wittmann, C. Biotechnology of riboflavin. Appl. Microbiol. Biotechnol. 2016, 100, 2107–2119. [Google Scholar] [CrossRef]
- Reihl, P.; Stolz, J. The Monocarboxylate Transporter Homolog Mch5p Catalyzes Riboflavin (Vitamin B2) Uptake inSaccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 39809–39817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, M.D.L.A.; Jiménez, A.; Revuelta, J. Molecular Characterization ofFMN1, the Structural Gene for the Monofunctional Flavokinase ofSaccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 28618–28624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Repetto, B.; Glerum, D.M.; Tzagoloff, A. Cloning and characterization of FAD1, the structural gene for flavin adenine dinucleotide synthetase of Saccharomyces cerevisiae. Mol. Cell. Biol. 1995, 15, 264–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frago, S.; Martínez-Júlvez, M.; Serrano, A.; Medina, M. Structural analysis of FAD synthetase from Corynebacterium ammoniagenes. BMC Microbiol. 2008, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Herguedas, B.; Martínez-Júlvez, M.; Frago, S.; Medina, M.; Hermoso, J.A. Oligomeric State in the Crystal Structure of Modular FAD Synthetase Provides Insights into Its Sequential Catalysis in Prokaryotes. J. Mol. Biol. 2010, 400, 218–230. [Google Scholar] [CrossRef]
- Sebastian, M.; Lira-Navarrete, E.; Serrano, A.; Marcuello, C.; Velazquez-Campoy, A.; Lostao, A.; Guerrero, R.H.; Medina, M.; Martínez-Júlvez, M. The FAD synthetase from the human pathogen Streptococcus pneumoniae: A bifunctional enzyme exhibiting activity-dependent redox requirements. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Leulliot, N.; Blondeau, K.; Keller, J.; Ulryck, N.; Quevillon-Cheruel, S.; Van Tilbeurgh, H. Crystal Structure of Yeast FAD Synthetase (Fad1) in Complex with FAD. J. Mol. Biol. 2010, 398, 641–646. [Google Scholar] [CrossRef]
- Sebastian, M.; Anoz-Carbonell, E.; Gracia, B.; Cossio, P.; Aínsa, J.A.; Lans, I.; Medina, M. Discovery of antimicrobial compounds targeting bacterial type FAD synthetases. J. Enzym. Inhib. Med. Chem. 2017, 33, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Locato, V.; Barile, M. A Regulatory Role of NAD Redox Status on Flavin Cofactor Homeostasis inS. cerevisiaeMitochondria. Oxidative Med. Cell. Longev. 2013, 2013, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giancaspero, T.A.; Dipalo, E.; Miccolis, A.; Boles, E.; Caselle, M.; Barile, M. Alteration of ROS Homeostasis and Decreased Lifespan inS. cerevisiaeElicited by Deletion of the Mitochondrial TranslocatorFLX1. BioMed Res. Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dufay, J.N.; Fernández-Murray, J.P.; McMaster, C.R. SLC25 Family Member Genetic Interactions Identify a Role for HEM25 in Yeast Electron Transport Chain Stability. G3: Genes|Genomes|Genetics 2017, 7, 1861–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toplak, M.; Brunner, J.; Tabib, C.R.; Macheroux, P. Closing the gap: Yeast electron-transferring flavoprotein links the oxidation of d-lactate and d-α-hydroxyglutarate to energy production via the respiratory chain. FEBS J. 2019, 286, 3611–3628. [Google Scholar] [CrossRef]
- Lopes, J.; Pinto, M.J.; Rodrigues, A.; Vasconcelos, F.; Oliveira, R. The Saccharomyces cerevisiae Genes, AIM45, YGR207c/CIR1 and YOR356w/CIR2, Are Involved in Cellular Redox State Under Stress Conditions. Open Microbiol. J. 2010, 4, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.-X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.-P.; Kunst, H.; Devilee, P.; Cremers, C.W.R.J.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, Is Mutated in Paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Maklashina, E.; Rajagukguk, S.; Iverson, T.M.; Cecchini, G. The unassembled flavoprotein subunits of human and bacterial complex II have impaired catalytic activity and generate only minor amounts of ROS. J. Biol. Chem. 2018, 293, 7754–7765. [Google Scholar] [CrossRef] [Green Version]
- Wanders, R.J.A.; Van Roermund, C.W.; Visser, W.F.; Ferdinandusse, S.; Jansen, G.A.; Brink, D.M.V.D.; Gloerich, J.; Waterham, H.R. Peroxisomal fatty acid alpha- and beta-oxidation in health and disease: New insights. Retin. Degener. Dis. 2003, 544, 293–302. [Google Scholar] [CrossRef]
- Byerly, L.; Cassada, R.; Russell, R. The life cycle of the nematode Caenorhabditis elegans. Dev. Biol. 1976, 51, 23–33. [Google Scholar] [CrossRef]
- The C. Elegans Sequencing Consortium. Elegans Sequencing Consortium. Genome Sequence of the Nematode, C. elegans: A Platform for Investigating Biology. Science 1998, 282, 2012–2018. [Google Scholar] [CrossRef]
- Biswas, A.; Elmatari, D.; Rothman, J.A.; LaMunyon, C.W.; Said, H.M. Identification and Functional Characterization of the Caenorhabditis elegans Riboflavin Transporters rft-1 and rft-2. PLOS ONE 2013, 8, e58190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhimathi, K.; Karthi, S.; Manimaran, P.; Varalakshmi, P.; AshokKumar, B. Riboflavin transporter-2 (rft-2) of Caenorhabditis elegans: Adaptive and developmental regulation. J. Biosci. 2015, 40, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a "good" look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.K.; Cornelius, N.; Gregersen, N. Genetic and cellular modifiers of oxidative stress: What can we learn from fatty acid oxidation defects? Mol. Genet. Metab. 2013, 110, S31–S39. [Google Scholar] [CrossRef]
- Qi, B.; Kniazeva, M.; Han, M. A vitamin-B2-sensing mechanism that regulates gut protease activity to impact animal’s food behavior and growth. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Yoshimatsu, H.; Yonezawa, A.; Yamanishi, K.; Yao, Y.; Sugano, K.; Nakagawa, S.; Imai, S.; Omura, T.; Nakagawa, T.; Yano, I.; et al. Disruption of Slc52a3 gene causes neonatal lethality with riboflavin deficiency in mice. Sci. Rep. 2016, 6, 27557. [Google Scholar] [CrossRef]
- Intoh, A.; Suzuki, N.; Koszka, K.; Eggan, K. SLC52A3, A Brown–Vialetto–van Laere syndrome candidate gene is essential for mouse development, but dispensable for motor neuron differentiation. Hum. Mol. Genet. 2016, 25, 1814–1823. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, V.S.; Lambrecht, N.W.G.; Lytle, C.; Said, H.M. Conditional (intestinal-specific) knockout of the riboflavin transporter-3 (RFVT-3) impairs riboflavin absorption. Am. J. Physiol. Liver Physiol. 2016, 310, G285–G293. [Google Scholar] [CrossRef] [Green Version]
- Chew, D.; Mah, A.K.; Baillie, D.L. Characterizing the transcriptional regulation of let-721, a Caenorhabditis elegans homolog of human electron flavoprotein dehydrogenase. Mol. Genet. Genom. 2009, 282, 555–570. [Google Scholar] [CrossRef]
- Alves, E.; Henriques, B.; Rodrigues, J.V.; Prudêncio, P.; Rocha, H.; Vilarinho, L.; Martinho, R.G.; Gomes, C.M. Mutations at the flavin binding site of ETF:QO yield a MADD-like severe phenotype in Drosophila. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2012, 1822, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Selak, M.A.; Watson, C.T.; Coutts, C.; Scherer, P.C.; Panzer, J.A.; Gibbs, S.; Scott, M.O.; Willer, G.; Gregg, R.G.; et al. Mechanisms Underlying Metabolic and Neural Defects in Zebrafish and Human Multiple Acyl-CoA Dehydrogenase Deficiency (MADD). PLOS ONE 2009, 4, e8329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-H.; Scott, S.A.; Bennett, M.J.; Carson, R.P.; Fessel, J.; Brown, H.A.; Ess, K.C. Multi-organ Abnormalities and mTORC1 Activation in Zebrafish Model of Multiple Acyl-CoA Dehydrogenase Deficiency. PLoS Genet. 2013, 9, e1003563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Li, D.; Lv, J.; Xu, X.; Wen, B.; Lin, P.; Liu, F.; Ji, K.; Shan, J.; Li, H.; et al. ETFDH Mutations and Flavin Adenine Dinucleotide Homeostasis Disturbance Are Essential for Developing Riboflavin-Responsive Multiple Acyl–Coenzyme A Dehydrogenation Deficiency. Ann. Neurol. 2018, 84, 659–673. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolomeo, M.; Nisco, A.; Leone, P.; Barile, M. Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy. Int. J. Mol. Sci. 2020, 21, 5310. https://doi.org/10.3390/ijms21155310
Tolomeo M, Nisco A, Leone P, Barile M. Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy. International Journal of Molecular Sciences. 2020; 21(15):5310. https://doi.org/10.3390/ijms21155310
Chicago/Turabian StyleTolomeo, Maria, Alessia Nisco, Piero Leone, and Maria Barile. 2020. "Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy" International Journal of Molecular Sciences 21, no. 15: 5310. https://doi.org/10.3390/ijms21155310
APA StyleTolomeo, M., Nisco, A., Leone, P., & Barile, M. (2020). Development of Novel Experimental Models to Study Flavoproteome Alterations in Human Neuromuscular Diseases: The Effect of Rf Therapy. International Journal of Molecular Sciences, 21(15), 5310. https://doi.org/10.3390/ijms21155310