NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis




Abstract
:1. Acute Pancreatitis: Initiating Events and Disease Progression
Pattern Recognition Receptors Contributing to Inflammation in Acute Pancreatitis
2. Inflammasomes
2.1. The NLRP3 Inflammasome
2.2. Signals of Action of the NLRP3 Inflammasome in the Immune Response
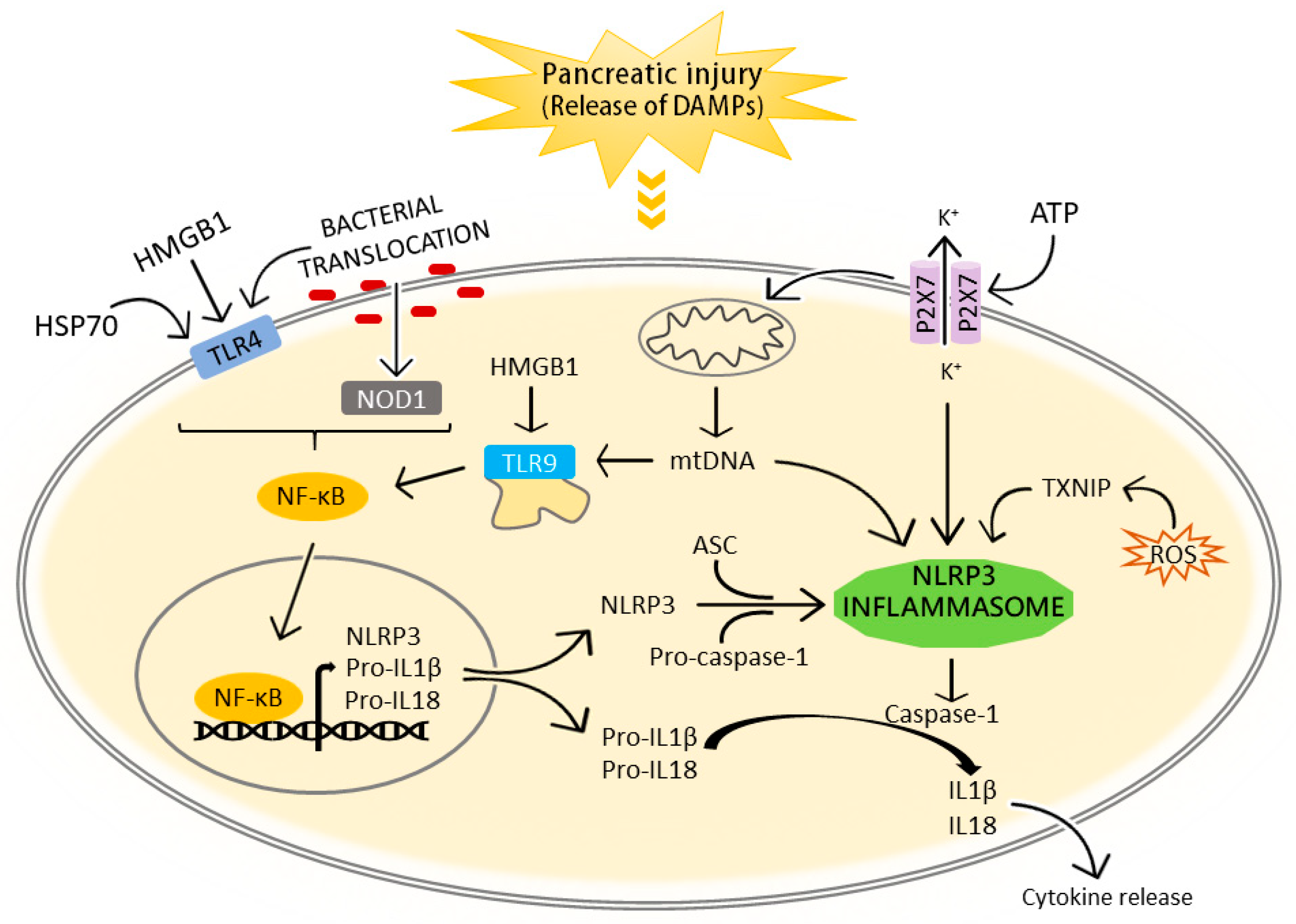
3. The NLRP3 Inflammasome in the Pathogenesis of Acute Pancreatitis and Associated Lung Injury
3.1. NLRP3 Inflammasome Activation in Acute Pancreatitis
3.1.1. Extracellular DAMPs
3.1.2. Bacterial Translocation
3.2. The IL1 Family of Cytokines as Effectors of the NLRP3 Inflammasome: Their Role in Acute Pancreatitis
3.2.1. IL1β
3.2.2. IL18
3.2.3. IL33
4. Inhibitors of the NLRP3 Inflammasome
4.1. The Clinical Application of NLRP3 Inflammasome Inhibitors: the IL1 Antagonists
4.2. NLRP3 Inflammasome Inhibitors in Acute Pancreatitis
4.2.1. Sulphonylureas Drugs
4.2.2. Natural Products from Plants and Fungi
4.2.3. Non-Steroidal Anti-Inflammatory Drugs and Other Antioxidants
4.2.4. Bile Acids
4.2.5. Antibiotics
5. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIM | Absence in melanoma 2 |
| ALRs | Absence in melanoma 2-like receptors |
| AP | Acute pancreatitis |
| ASC | Caspase recruitment domain |
| CAPS | Cryopyrin-associated periodic syndrome |
| CARD | Caspase activation and recruitment domain |
| CARS | Compensatory anti-inflammatory response syndrome |
| CLRs | C-type lectin receptors |
| COX-2 | Cyclo-oxygenase-2 |
| DAMPs | Damage-Associated Molecular Patterns |
| GSDM-D | Gasdermin-D |
| HMGB1 | High mobility group box 1 |
| IL | Interleukin |
| IL1R | Interleukin 1 receptor |
| IRAK | IL1R-associated kinase |
| IRF | Interferon regulatory factor |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MyD88 | Myeloid differentiation primary response 88 |
| NATCH | Nucleotide-binding and oligomerization domain |
| NF-κB | Nuclear factor-kappa B |
| NLRP3 | NLR pyrin domain containing protein 3 |
| NOD1 | NLR nucleotide-binding oligomerization domain-containing protein 1 |
| Nrf2 | Nuclear factor erythrocyte-2 associated factor 2 |
| NSAIDS | Non-steroidal anti-inflammatory drugs |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PRR | Pattern recognition receptors |
| PYD | Pyrin domain |
| RAGE | Receptor for advanced glycation end-products |
| RLRs | Retinoic acid-inducible gene (RIG)-I-like receptors |
| ROS | Reactive oxygen species |
| SIRS | Systemic inflammatory response syndrome |
| TLR | Toll-like receptor |
| TNFα | Tumor necrosis factor alpha |
| TRAF | Tumor necrosis factor receptor-activated factor |
| TRIF | TIR-domain-containing adaptor molecule inducing interferon-beta |
References
- Dumnicka, P.; Maduzia, D.; Ceranowicz, P.; Olszanecki, R.; Drożdż, R.; Kuśnierz-Cabala, B. The interplay between inflammation, coagulation and endothelial injury in the early phase of acute pancreatitis: Clinical implications. Int. J. Mol. Sci. 2017, 18, 354. [Google Scholar] [CrossRef] [Green Version]
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S. Classification of acute pancreatitis-2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Zhou, M.T.; Chen, C.S.; Chen, B.C.; Zhang, Q.Y.; Andersson, R. Acute lung injury and ARDS in acute pancreatitis: Mechanisms and potential intervention. World J. Gastroenterol. 2010, 16, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.L.; DiSario, J.A.; Nelson, D.B.; Fennerty, M.B.; Lee, J.G.; Bjorkman, D.J.; Overby, C.S.; Aas, J.; Ryan, M.E.; Bochna, G.S.; et al. Risk factors for post-ERCP pancreatitis: A prospective, multicenter study. Gastrointest. Endosc. 2001, 54, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.W.; Meng, X.X.; Xu, P. Central role of neutrophil in the pathogenesis of severe acute pancreatitis. J. Cell. Mol. Med. 2015, 19, 2513–2520. [Google Scholar] [CrossRef]
- Janiak, A.; Leśniowski, B.; Jasińska, A.; Pietruczuk, M.; Małecka-Panas, E. Interleukin 18 as an early marker or prognostic factor in acute pancreatitis. Prz. Gastroenterol. 2015, 10, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Sharif, R.; Dawra, R.; Wasiluk, K.; Phillips, P.; Dudeja, V.; Kurt-Jones, E.; Finberg, R.; Saluja, A. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut 2009, 58, 813–819. [Google Scholar] [CrossRef]
- Pastor, C.M.; Pugin, J.; Kwak, B.; Chanson, M.; Mach, F.; Hadengue, A.; Frossard, J.L. Role of Toll-like receptor 4 on pancreatic and pulmonary injury in a mice model of acute pancreatitis associated with endotoxemia. Crit. Care Med. 2004, 32, 1759–1763. [Google Scholar] [CrossRef]
- Sawa, H.; Ueda, T.; Takeyama, Y.; Yasuda, T.; Shinzeki, M.; Nakajima, T.; Kuroda, Y. Role of toll-like receptor 4 in the pathophysiology of severe acute pancreatitis in mice. Surg. Today 2007, 37, 867–873. [Google Scholar] [CrossRef]
- Hoque, R.; Sohail, M.; Malik, A.; Sarwar, S.; Luo, Y.; Shah, A.; Barrat, F.; Flavell, R.; Gorelick, F.; Husain, S.; et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 2011, 141, 358–369. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, Y.; Watanabe, T.; Kudo, M.; Arai, H.; Strober, W.; Chiba, T. Sensing of commensal organisms by the intracellular sensor NOD1 mediates experimental pancreatitis. Immunity 2012, 37, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Chen, R.; Xie, M.; Cao, L.; Lotze, M.T.; Tang, D.; Zeh, H.J. The receptor for advanced glycation end products activates the AIM2 inflammasome in acute pancreatitis. J. Immunol. 2016, 196, 4331–4337. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Mason, D.R.; Beck, P.L.; Muruve, D.A. Nucleotide-binding oligomerization domain-like receptors and inflammasomes in the pathogenesis of non-microbial inflammation and diseases. J. Innate Immun. 2012, 4, 16–30. [Google Scholar] [CrossRef]
- Kummer, J.A.; Broekhuizen, R.; Everett, H.; Agostini, L.; Kuijk, L.; Martinon, F.; van Bruggen, R.; Tschopp, J. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J. Histochem. Cytochem. J. Histochem. Soc. 2007, 55, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Castejon, G.; Luheshi, N.M.; Compan, V.; High, S.; Whitehead, R.C.; Flitsch, S.; Kirov, A.; Prudovsky, I.; Swanton, E.; Brough, D. Deubiquitinases regulate the activity of caspase-1 and interleukin-1β secretion via assembly of the inflammasome. J. Biol. Chem. 2013, 288, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol. Cell 2017, 68, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Barry, R.; John, S.W.; Liccardi, G.; Tenev, T.; Jaco, I.; Chen, C.H.; Choi, J.; Kasperkiewicz, P.; Fernandes-Alnemri, T.; Alnemri, E.; et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat. Commun. 2018, 9, 3001. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Segovia, J.A.; Somarajan, S.R.; Chang, T.H.; Kannan, T.R.; Baseman, J.B. ADP-ribosylation of NLRP3 by mycoplasma pneumoniae CARDS toxin regulates inflammasome activity. mBio 2014, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Wang, H.; Andersson, U.; Tracey, K.J. Regulation of HMGB1 release by inflammasomes. Protein Cell 2013, 4, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front. Immunol. 2017, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Malmstrøm, M.L.; Hansen, M.B.; Andersen, A.M.; Ersbøll, A.K.; Nielsen, O.H.; Jørgensen, L.N.; Novovic, S. Cytokines and organ failure in acute pancreatitis: Inflammatory response in acute pancreatitis. Pancreas 2012, 41, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Rau, B.; Gansauge, F.; Beger, H.G. Inflammatory mediators in human acute pancreatitis: Clinical and pathophysiological implications. Gut 2000, 47, 546–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Zhai, Z.; Wang, Y.; Xu, L.; Jia, P.; Xia, P.; Liu, C.; Zhang, X.; Qin, T.; Zhang, H. NLRP3 deficiency alleviates severe acute pancreatitis and pancreatitis-associated lung injury in a mouse model. BioMed Res. Int. 2018, 2018, 1294951. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Farooq, A.; Ghani, A.; Gorelick, F.; Mehal, W.Z. Lactate reduces liver and pancreatic injury in toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 2014, 146, 1763–1774. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.U.; Hwang, J.Q.; Gardner, T.H.; Repas, K.; Delee, R.; Yu, S.; Smith, B.; Banks, P.A.; Conwell, D.L. Lactated ringer’s solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin. Gastroenterol. Hepatol. Clin. Pract. J. Am. Gastroenterol. Assoc. 2011, 9, 710–717.e1. [Google Scholar] [CrossRef]
- Madaria, D.E.; Herrera-Marante, I.; González-Camacho, V.; Bonjoch, L.; Quesada-Vázquez, N.; Almenta-Saavedra, I.; Miralles-Maciá, C.; Acevedo-Piedra, N.G.; Roger-Ibáñez, M.; Sánchez-Marin, C.; et al. Fluid resuscitation with lactated Ringer’s solution vs normal saline in acute pancreatitis: A triple-blind, randomized, controlled trial. United Eur. Gastroenterol. J. 2018, 6, 63–72. [Google Scholar] [CrossRef]
- Shen, A.; Kim, H.J.; Oh, G.S.; Lee, S.B.; Lee, S.H.; Pandit, A.; Khadka, D.; Choe, S.K.; Kwak, S.C.; Yang, S.H.; et al. NAD(+) augmentation ameliorates acute pancreatitis through regulation of inflammasome signalling. Sci. Rep. 2017, 7, 3006. [Google Scholar] [CrossRef]
- Xue, J.; Habtezion, A. Carbon monoxide-based therapy ameliorates acute pancreatitis via TLR4 inhibition. J. Clin. Investig. 2014, 124, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.B.; Sun, H.Y.; Luo, Z.L.; Cheng, L.; Duan, X.M.; Ren, J.D. Plasma-derived exosomes contribute to pancreatitis-associated lung injury by triggering NLRP3-dependent pyroptosis in alveolar macrophages. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165685. [Google Scholar] [CrossRef]
- Chueca, A.F.; Madaria, D.E.; Ruiz, L.B.; Cardona, M.C.; Vázquez, Q.N.; Bachiller, V.; Tarín, F.; Such, J.; Francés, R.; Zapater, P.; et al. The expression and activation of the AIM2 inflammasome correlates with inflammation and disease severity in patients with acute pancreatitis. Pancreatol. J. Int. Assoc. Pancreatol. 2017, 17, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Sendler, M.; Van den Brandt, C.; Glaubitz, J.; Wilden, A.; Golchert, J.; Weiss, F.U.; Homuth, G.; De Freitas Chama, L.L.; Mishra, N.; Mahajan, U.M.; et al. NLRP3 inflammasome regulates development of systemic inflammatory response and compensatory anti-inflammatory response syndromes in mice with acute pancreatitis. Gastroenterology 2020, 158, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calogero, S.; Grassi, F.; Aguzzi, A.; Voigtländer, T.; Ferrier, P.; Ferrari, S.; Bianchi, M.E. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat. Genet. 1999, 22, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, W.Q. High-mobility group box 1 protein and its role in severe acute pancreatitis. World J. Gastroenterol. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Yasuda, T.; Ueda, T.; Takeyama, Y.; Shinzeki, M.; Sawa, H.; Nakajima, T.; Ajiki, T.; Fujino, Y.; Suzuki, Y.; Kuroda, Y. Significant increase of serum high-mobility group box chromosomal protein 1 levels in patients with severe acute pancreatitis. Pancreas 2006, 33, 359–363. [Google Scholar] [CrossRef]
- Kocsis, A.K.; Szabolcs, A.; Hofner, P.; Takács, T.; Farkas, G.; Boda, K.; Mándi, Y. Plasma concentrations of high-mobility group box protein 1, soluble receptor for advanced glycation end-products and circulating DNA in patients with acute pancreatitis. Pancreatol. J. Int. Assoc. Pancreatol. 2009, 9, 383–391. [Google Scholar] [CrossRef]
- Lindström, O.; Tukiainen, E.; Kylänpää, L.; Mentula, P.; Rouhiainen, A.; Puolakkainen, P.; Rauvala, H.; Repo, H. Circulating levels of a soluble form of receptor for advanced glycation end products and high-mobility group box chromosomal protein 1 in patients with acute pancreatitis. Pancreas 2009, 38, 215–220. [Google Scholar] [CrossRef]
- Yasuda, T.; Ueda, T.; Shinzeki, M.; Sawa, H.; Nakajima, T.; Takeyama, Y.; Kuroda, Y. Increase of high-mobility group box chromosomal protein 1 in blood and injured organs in experimental severe acute pancreatitis. Pancreas 2007, 34, 487–488. [Google Scholar] [CrossRef]
- Sawa, H.; Ueda, T.; Takeyama, Y.; Yasuda, T.; Shinzeki, M.; Nakajima, T.; Kuroda, Y. Blockade of high mobility group box-1 protein attenuates experimental severe acute pancreatitis. World J. Gastroenterol. 2006, 12, 7666–7670. [Google Scholar] [CrossRef]
- Luan, Z.G.; Zhang, X.J.; Yin, X.H.; Ma, X.C.; Zhang, H.; Zhang, C.; Guo, R.X. Downregulation of HMGB1 protects against the development of acute lung injury after severe acute pancreatitis. Immunobiology 2013, 218, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Jin, X.; Sun, J.; Li, F.; Feng, Q.; Zhang, C.; Cao, Y.; Wang, Y. Protective effect of HMGB1 a box on organ injury of acute pancreatitis in mice. Pancreas 2009, 38, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.W.; Zhang, Q.Y.; Zhou, M.T.; Liu, N.X.; Chen, T.K.; Zhu, Y.F.; Wu, L. Antioxidant inhibits HMGB1 expression and reduces pancreas injury in rats with severe acute pancreatitis. Dig. Dis. Sci. 2010, 55, 2529–2536. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Ling, Y.; Yin, T.; Tao, J.; Xiong, J.X.; Wu, H.S.; Wang, C.Y. Delayed ethyl pyruvate therapy attenuates experimental severe acute pancreatitis via reduced serum high mobility group box 1 levels in rats. World J. Gastroenterol. 2008, 14, 4546–4550. [Google Scholar] [CrossRef]
- Chen, R.; Hou, W.; Zhang, Q.; Kang, R.; Fan, X.G.; Tang, D. Emerging role of high-mobility group box 1 (HMGB1) in liver diseases. Mol. Med. Camb. Mass. 2013, 19, 357–366. [Google Scholar] [CrossRef]
- Li, G.; Wu, X.; Yang, L.; He, Y.; Liu, Y.; Jin, X.; Yuan, H. TLR4-mediated NF-κB signaling pathway mediates HMGB1-induced pancreatic injury in mice with severe acute pancreatitis. Int. J. Mol. Med. 2016, 37, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Yanai, H.; Ban, T.; Wang, Z.; Choi, M.K.; Kawamura, T.; Negishi, H.; Nakasato, M.; Lu, Y.; Hangai, S.; Koshiba, R.; et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 2009, 462, 99–103. [Google Scholar] [CrossRef]
- Nover, L.; Scharf, K.D. Heat stress proteins and transcription factors. Cell. Mol. Life Sci. 1997, 53, 80–103. [Google Scholar] [CrossRef]
- Pilon, M.; Schekman, R. Protein translocation: How Hsp70 pulls it off. Cell 1999, 97, 679–682. [Google Scholar] [CrossRef] [Green Version]
- Singh-Jasuja, H.; Hilf, N.; Arnold-Schild, D.; Schild, H. The role of heat shock proteins and their receptors in the activation of the immune system. Biol. Chem. 2001, 382, 629–636. [Google Scholar] [CrossRef]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakonczay, J.Z.; Takács, T.; Boros, I.; Lonovics, J. Heat shock proteins and the pancreas. J. Cell. Physiol. 2003, 195, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Balog, A.; Gyulai, Z.; Boros, L.G.; Farkas, G.; Takács, T.; Lonovics, J.; Mándi, Y. Polymorphism of the TNF-alpha, HSP70-2, and CD14 genes increases susceptibility to severe acute pancreatitis. Pancreas 2005, 30, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Song, J.M.; Liu, H.X.; Li, Y.; Zeng, Y.J.; Zhou, Z.G.; Liu, H.Y.; Xu, B.; Wang, L.; Zhou, B.; Wang, R. Extracellular heat-shock protein 70 aggravates cerulein-induced pancreatitis through toll-like receptor-4 in mice. Chin. Med. J. 2008, 121, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Somensi, N.; Brum, P.O.; Miranda, d.R.V.; Gasparotto, J.; Filho, Z.A.; Rostirolla, D.C.; Silva, d.M.M.; Moreira, J.C.F.; Gelain, P.D. Extracellular HSP70 activates ERK1/2, NF-kB and pro-inflammatory gene transcription through binding with RAGE in A549 human lung cancer cells. Cell. Physiol. Biochem. 2017, 42, 2507–2522. [Google Scholar] [CrossRef]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.; Cheema, H.; George, J.; Iyer, S.; Dudeja, V.; Dawra, R.; Saluja, A.K. Extracellular release of ATP promotes systemic inflammation during acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, 463–475. [Google Scholar] [CrossRef]
- Frossard, J.L.; Steer, M.L.; Pastor, C.M. Acute pancreatitis. Lancet Lond. Engl. 2008, 371, 143–152. [Google Scholar] [CrossRef]
- Mithöfer, K.; Fernández, -d.C.C.; Ferraro, M.J.; Lewandrowski, K.; Rattner, D.W.; Warshaw, A.L. Antibiotic treatment improves survival in experimental acute necrotizing pancreatitis. Gastroenterology 1996, 110, 232–240. [Google Scholar]
- Fritz, S.; Hartwig, W.; Lehmann, R.; Will-Schweiger, K.; Kommerell, M.; Hackert, T.; Schneider, L.; Büchler, M.W.; Werner, J. Prophylactic antibiotic treatment is superior to therapy on-demand in experimental necrotising pancreatitis. Crit. Care Lond. Engl. 2008, 12, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foitzik, T.; Fernández, -d.C.C.; Ferraro, M.J.; Mithöfer, K.; Rattner, D.W.; Warshaw, A.L. Pathogenesis and prevention of early pancreatic infection in experimental acute necrotizing pancreatitis. Ann. Surg. 1995, 222, 179–185. [Google Scholar] [CrossRef]
- Luiten, E.J.; Hop, W.C.; Endtz, H.P.; Bruining, H.A. Prognostic importance of gram-negative intestinal colonization preceding pancreatic infection in severe acute pancreatitis. Results of a controlled clinical trial of selective decontamination. Intensive Care Med. 1998, 24, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Luiten, E.J.; Hop, W.C.; Lange, J.F.; Bruining, H.A. Controlled clinical trial of selective decontamination for the treatment of severe acute pancreatitis. Ann. Surg. 1995, 222, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. IL-1: Discoveries, controversies and future directions. Eur. J. Immunol. 2010, 40, 599–606. [Google Scholar] [CrossRef]
- Lukens, J.R.; Gross, J.M.; Kanneganti, T.D. IL-1 family cytokines trigger sterile inflammatory disease. Front. Immunol. 2012, 3, 315. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Kudo, M.; Strober, W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017, 10, 283–298. [Google Scholar] [CrossRef] [Green Version]
- Hoque, R.; Mehal, W.Z. Inflammasomes in pancreatic physiology and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A.; Simon, A.; Meer, v.d.J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; van de Veerdonk, F.L.; Meer, v.d.J.W.; Dinarello, C.A.; Joosten, L.A. Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol. 2015, 33, 49–77. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural basis of IL-1 family cytokine signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Bond, J.S. Prointerleukin-18 is activated by meprin beta in vitro and in vivo in intestinal inflammation. J. Biol. Chem. 2008, 283, 31371–31377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, T.; Takeyama, Y.; Yasuda, T.; Matsumura, N.; Sawa, H.; Nakajima, T.; Ajiki, T.; Fujino, Y.; Suzuki, Y.; Kuroda, Y. Significant elevation of serum interleukin-18 levels in patients with acute pancreatitis. J. Gastroenterol. 2006, 41, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Siemiatkowska, W.U.; Mroczko, B.; Siemiatkowski, A. Serum profiles of interleukin-18 in different severity forms of human acute pancreatitis. Scand. J. Gastroenterol. 2002, 37, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Rau, B.; Baumgart, K.; Paszkowski, A.S.; Mayer, J.M.; Beger, H.G. Clinical relevance of caspase-1 activated cytokines in acute pancreatitis: High correlation of serum interleukin-18 with pancreatic necrosis and systemic complications. Crit. Care Med. 2001, 29, 1556–1562. [Google Scholar] [CrossRef]
- Pastor, C.M.; Morel, D.R.; Vonlaufen, A.; Schiffer, E.; Lescuyer, P.; Frossard, J.L. Delayed production of IL-18 in lungs and pancreas of rats with acute pancreatitis. Pancreatol. J. Int. Assoc. Pancreatol. 2010, 10, 752–757. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zhu, R.M.; Xu, W.A.; Wan, H.J.; Lu, H. Therapeutic effects of caspase-1 inhibitors on acute lung injury in experimental severe acute pancreatitis. World J. Gastroenterol. 2007, 13, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Ueno, N.; Kashiwamura, S.; Ueda, H.; Okamura, H.; Tsuji, N.M.; Hosohara, K.; Kotani, J.; Marukawa, S. Role of interleukin 18 in nitric oxide production and pancreatic damage during acute pancreatitis. Shock 2005, 24, 564–570. [Google Scholar] [CrossRef]
- Sennello, J.A.; Fayad, R.; Pini, M.; Gove, M.E.; Ponemone, V.; Cabay, R.J.; Siegmund, B.; Dinarello, C.A.; Fantuzzi, G. Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 8085–8090. [Google Scholar] [CrossRef] [Green Version]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Lefrançais, E.; Roga, S.; Gautier, V.; Peredo, G.d.A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar]
- Pinto, S.M.; Subbannayya, Y.; Rex, D.A.B.; Raju, R.; Chatterjee, O.; Advani, J.; Radhakrishnan, A.; Keshava Prasad, T.S.; Wani, M.R.; Pandey, A. A network map of IL-33 signaling pathway. J. Cell Commun. Signal. 2018, 12, 615–624. [Google Scholar] [CrossRef]
- Kotsiou, O.S.; Gourgoulianis, K.I.; Zarogiannis, S.G. IL-33/ST2 axis in organ fibrosis. Front. Immunol. 2018, 9, 2432. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; An, Y.; Jiang, D.; Wu, B.; Yang, Y.; Sun, D. TNF-α regulating interleukin-33 induces acute pancreatic inflammation in rats. Ann. Clin. Lab. Sci. 2016, 46, 54–59. [Google Scholar]
- Xiang, H.; Tao, X.; Xia, S.; Qu, J.; Song, H.; Liu, J.; Shang, D. Emodin alleviates sodium taurocholate-induced pancreatic acinar cell injury via MicroRNA-30a-5p-mediated inhibition of high-temperature requirement a/transforming growth factor beta 1 inflammatory signaling. Front. Immunol. 2017, 8, 1488. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Twait, E.C.; Williard, D.E.; Yuan, Z.; Meyerholz, D.K.; Samuel, I. The novel cytokine interleukin-33 activates acinar cell proinflammatory pathways and induces acute pancreatic inflammation in mice. PLoS ONE 2013, 8, e56866. [Google Scholar] [CrossRef] [Green Version]
- Ouziel, R.; Gustot, T.; Moreno, C.; Arvanitakis, M.; Degré, D.; Trépo, E.; Quertinmont, E.; Vercruysse, V.; Demetter, P.; Le Moine, O.; et al. The ST2 pathway is involved in acute pancreatitis: A translational study in humans and mice. Am. J. Pathol. 2012, 180, 2330–2339. [Google Scholar] [CrossRef]
- Sesti-Costa, R.; Silva, G.K.; Módena, P.J.L.; Carlos, D.; Silva, M.L.; Alves-Filho, J.C.; Arruda, E.; Liew, F.Y.; Silva, J.S. The IL-33/ST2 pathway controls coxsackievirus B5-induced experimental pancreatitis. J. Immunol. 2013, 191, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.; Yazgan, Y.; Tanoglu, A.; Berber, U.; Oncu, K.; Kara, M.; Demirel, D.; Kucuk, I.; Ozari, H.O.; Ipcioglu, O.M. Effectiveness of interleukin-1 receptor antagonist (Anakinra) on cerulein-induced experimental acute pancreatitis in rats. Scand. J. Gastroenterol. 2014, 49, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Throne, M.L.; Amar, N.J.; Sebai, M.; Kivitz, A.J.; Kavanaugh, A.; Weinstein, S.P.; Belomestnov, P.; Yancopoulos, G.D.; Stahl, N.; et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheum. 2008, 58, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.R.; McGonagle, D.; Nizam, S.; Jarrett, S.; Hilst, v.d.J.; McDermott, M.F.; Savic, S. Anakinra as a diagnostic challenge and treatment option for systemic autoinflammatory disorders of undefined etiology. JCI Insight 2016, 1, 86336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, N.; Radin, A.; Mellis, S. Rilonacept--CAPS and beyond. Ann. N. Y. Acad. Sci. 2009, 1182, 124–134. [Google Scholar] [CrossRef]
- Rondeau, J.M.; Ramage, P.; Zurini, M.; Gram, H. The molecular mode of action and species specificity of canakinumab, a human monoclonal antibody neutralizing IL-1β. mAbs 2015, 7, 1151–1160. [Google Scholar] [CrossRef] [Green Version]
- Fenini, G.; Contassot, E.; French, L.E. Potential of IL-1, IL-18 and inflammasome inhibition for the treatment of inflammatory skin diseases. Front. Pharmacol. 2017, 8, 278. [Google Scholar] [CrossRef]
- Landmann, E.C.; Walker, U.A. Pharmacological treatment options for cryopyrin-associated periodic syndromes. Expert Rev. Clin. Pharmacol. 2017, 10, 855–864. [Google Scholar] [CrossRef]
- York, J.M.; Castellanos, K.J.; Cabay, R.J.; Fantuzzi, G. Inhibition of the nucleotide-binding domain, leucine-rich containing family, pyrin-domain containing 3 inflammasome reduces the severity of experimentally induced acute pancreatitis in obese mice. Transl. Res. J. Lab. Clin. Med. 2014, 164, 259–269. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Tao, X.; Xia, S.; Qu, J.; Song, H.; Liu, J.; Li, H.; Shang, D. Emodin attenuated severe acute pancreatitis via the P2X ligand-gated ion channel7/NOD-like receptor protein 3 signaling pathway. Oncol. Rep. 2019, 41, 270–278. [Google Scholar]
- Gao, Z.; Sui, J.; Fan, R.; Qu, W.; Dong, X.; Sun, D. Emodin protects against acute pancreatitis-associated lung injury by inhibiting NLPR3 inflammasome activation via Nrf2/HO-1 signaling. Drug Des. Dev. Ther. 2020, 14, 1971–1982. [Google Scholar] [CrossRef]
- Ren, Z.; Li, H.; Zhang, M.; Zhao, Y.; Fang, X.; Li, X.; Chen, W.; Zhang, H.; Wang, Y.; Pan, L.L.; et al. A novel derivative of the natural product danshensu suppresses inflammatory responses to alleviate caerulein-induced acute pancreatitis. Front. Immunol. 2018, 9, 2513. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bae, G.S.; Jo, I.J.; Choi, S.B.; Kim, D.G.; Jung, H.J.; Song, H.J.; Park, S.J. Fraxinellone inhibits inflammatory cell infiltration during acute pancreatitis by suppressing inflammasome activation. Int. Immunopharmacol. 2019, 69, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kanak, M.A.; Shahbazov, R.; Yoshimatsu, G.; Levy, M.F.; Lawrence, M.C.; Naziruddin, B. A small molecule inhibitor of NFκB blocks ER stress and the NLRP3 inflammasome and prevents progression of pancreatitis. J. Gastroenterol. 2017, 52, 352–365. [Google Scholar] [CrossRef]
- Aruna, R.; Geetha, A.; Suguna, P. Rutin modulates ASC expression in NLRP3 inflammasome: A study in alcohol and cerulein-induced rat model of pancreatitis. Mol. Cell. Biochem. 2014, 396, 269–280. [Google Scholar] [CrossRef]
- Dong, Z.; Shang, H.; Chen, Y.Q.; Pan, L.L.; Bhatia, M.; Sun, J. Sulforaphane protects pancreatic acinar cell injury by modulating Nrf2-mediated oxidative stress and NLRP3 inflammatory pathway. Oxidative Med. Cell. Longev. 2016, 2016, 7864150. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, Y.; Shi, J. Cordycepin protects against acute pancreatitis by modulating NF-κB and NLRP3 inflammasome activation via AMPK. Life Sci. 2020, 251, 117645. [Google Scholar] [CrossRef]
- Lu, G.; Pan, Y.; Kayoumu, A.; Zhang, L.; Yin, T.; Tong, Z.; Li, B.; Xiao, W.; Ding, Y.; Li, W. Indomethacin inhabits the NLRP3 inflammasome pathway and protects severe acute pancreatitis in mice. Biochem. Biophys. Res. Commun. 2017, 493, 827–832. [Google Scholar] [CrossRef]
- Hou, C.; Zhu, X.; Shi, C.; Peng, Y.; Huang, D.; Li, Q.; Miao, Y. Iguratimod (T-614) attenuates severe acute pancreatitis by inhibiting the NLRP3 inflammasome and NF-κB pathway. Biomed. Pharmacother. 2019, 119, 109455. [Google Scholar] [CrossRef]
- Jin, H.Z.; Yang, X.J.; Zhao, K.L.; Mei, F.C.; Zhou, Y.; You, Y.D.; Wang, W.X. Apocynin alleviates lung injury by suppressing NLRP3 inflammasome activation and NF-κB signaling in acute pancreatitis. Int. Immunopharmacol. 2019, 75, 105821. [Google Scholar] [CrossRef]
- Li, B.; Yang, N.; Li, C.; Li, C.; Gao, K.; Xie, X.; Dong, X.; Yang, J.; Yang, Q.; Tong, Z.; et al. INT-777, a bile acid receptor agonist, extenuates pancreatic acinar cells necrosis in a mouse model of acute pancreatitis. Biochem. Biophys. Res. Commun. 2018, 503, 38–44. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological inhibitors of the NLRP3 inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [Green Version]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Ren, P.; Wu, D.; Appel, R.; Zhang, L.; Zhang, C.; Luo, W.; Robertson, A.A.B.; Cooper, M.A.; Coselli, J.S.; Milewicz, D.M.; et al. Targeting the NLRP3 inflammasome with inhibitor MCC950 prevents aortic aneurysms and dissections in mice. J. Am. Heart Assoc. 2020, 9, 014044. [Google Scholar] [CrossRef]
- Shrimali, D.; Shanmugam, M.K.; Kumar, A.P.; Zhang, J.; Tan, B.K.; Ahn, K.S.; Sethi, G. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013, 341, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.M.; Wang, F.Y.; Wang, Z.K.; Wan, H.J.; Xu, W.A.; Lu, H. Emodin enhances alveolar epithelial barrier function in rats with experimental acute pancreatitis. World J. Gastroenterol. 2010, 16, 2994–3001. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Huang, B.; Wang, Y.; Tong, C.; Xie, P.; Fan, R.; Gao, Z. Emodin ameliorates acute lung injury induced by severe acute pancreatitis through the up-regulated expressions of AQP1 and AQP5 in lung. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1071–1079. [Google Scholar] [CrossRef]
- Bao, X.Y.; Zheng, Q.; Tong, Q.; Zhu, P.C.; Zhuang, Z.; Zheng, G.Q.; Wang, Y. Danshensu for myocardial ischemic injury: Preclinical evidence and novel methodology of quality assessment tool. Front. Pharmacol. 2018, 9, 1445. [Google Scholar] [CrossRef]
- Yin, Y.; Duan, J.; Guo, C.; Wei, G.; Wang, Y.; Guan, Y.; Mu, F.; Yao, M.; Xi, M.; Wen, A. Danshensu accelerates angiogenesis after myocardial infarction in rats and promotes the functions of endothelial progenitor cells through SDF-1α/CXCR4 axis. Eur. J. Pharmacol. 2017, 814, 274–282. [Google Scholar] [CrossRef]
- Liu, X.H.; Pan, L.L.; Jia, Y.L.; Wu, D.; Xiong, Q.H.; Wang, Y.; Zhu, Y.Z. A novel compound DSC suppresses lipopolysaccharide-induced inflammatory responses by inhibition of Akt/NF-κB signalling in macrophages. Eur. J. Pharmacol. 2013, 708, 8–13. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, Y.M.; Shin, J.S.; Park, S.J.; Choi, J.H.; Jung, H.J.; Park, H.J.; Lee, K.T. Fraxinellone inhibits lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 expression by negatively regulating nuclear factor-kappa B in RAW 264.7 macrophages cells. Biol. Pharm. Bull. 2009, 32, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Mohan, R.; Hammers, H.J.; Bargagna-Mohan, P.; Zhan, X.H.; Herbstritt, C.J.; Ruiz, A.; Zhang, L.; Hanson, A.D.; Conner, B.P.; Rougas, J.; et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis 2004, 7, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Liu, J.; Feng, X.; Salazar Hernández, M.A.; Mucka, P.; Ibi, D.; Choi, J.W.; Ozcan, U. Withaferin A is a leptin sensitizer with strong antidiabetic properties in mice. Nat. Med. 2016, 22, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Park, J.M.; Jeong, M.; Han, Y.M.; Go, E.J.; Ko, W.J.; Cho, J.Y.; Kwon, C.I.; Hahm, K.B. Korean red ginseng ameliorated experimental pancreatitis through the inhibition of hydrogen sulfide in mice. Pancreatol. J. Int. Assoc. Pancreatol. 2016, 16, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Abreu, F.F.; Souza, A.C.; Teixeira, S.A.; Soares, A.G.; Teixeira, D.F.; Soares, R.C.; Santana, M.T.; Lauton Santos, S.; Costa, S.K.; Muscará, M.N.; et al. Elucidating the role of oxidative stress in the therapeutic effect of rutin on experimental acute pancreatitis. Free Radic. Res. 2016, 50, 1350–1360. [Google Scholar] [CrossRef]
- Liu, H.; Talalay, P. Relevance of anti-inflammatory and antioxidant activities of exemestane and synergism with sulforaphane for disease prevention. Proc. Natl. Acad. Sci. USA 2013, 110, 19065–19070. [Google Scholar] [CrossRef] [Green Version]
- Greaney, A.J.; Maier, N.K.; Leppla, S.H.; Moayeri, M. Sulforaphane inhibits multiple inflammasomes through an Nrf2-independent mechanism. J. Leukoc. Biol. 2016, 99, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.C.; Zhou, X.P.; Wang, X.A.; Xu, M.D.; Chen, T.; Chen, T.Y.; Zhou, P.H.; Zhang, Y.Q. Cordycepin induces apoptosis and G2/M phase arrest through the ERK pathways in esophageal cancer cells. J. Cancer 2019, 10, 2415–2424. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Chen, Z.; Jiang, Z.; Luo, P.; Liu, L.; Huang, Y.; Wang, H.; Wang, Y.; Long, L.; Tan, X.; et al. Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents. Nat. Commun. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Nallathamby, N.; Serm, G.L.; Vidyadaran, S.; Malek, A.S.N.; Raman, J.; Sabaratnam, V. Ergosterol of cordyceps militaris attenuates LPS induced inflammation in BV2 microglia cells. Nat. Prod. Commun. 2015, 10, 885–886. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Li, Y.Z.; Hylemon, P.B.; Zhang, L.Y.; Zhou, H.P. Cordycepin inhibits LPS-induced inflammatory responses by modulating NOD-Like Receptor Protein 3 inflammasome activation. Biomed. Pharmacother. 2017, 95, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Robles, L.; Vaziri, N.D.; Ichii, H. Role of oxidative stress in the pathogenesis of pancreatitis: Effect of antioxidant therapy. Pancreat. Disord. Ther. 2013, 3, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, K.F.; Chou, J.C.; Ka, S.M.; Tasi, Y.L.; Chen, A.; Wu, S.H.; Chiu, H.W.; Wong, W.T.; Wang, Y.F.; Tsai, C.L.; et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1β production. J. Cell. Physiol. 2015, 230, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Kornasoff, D.; Maisenbacher, J.; Bowdler, J.; Raber, A. The efficacy and tolerability of aceclofenac compared to indomethacin in patients with rheumatoid arthritis. Rheumatol. Int. 1996, 15, 225–230. [Google Scholar] [CrossRef]
- Nalamachu, S.; Wortmann, R. Role of indomethacin in acute pain and inflammation management: A review of the literature. Postgrad. Med. 2014, 126, 92–97. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, L.; Leung, J.; Zhang, R.; Liu, Z.; Wang, X.; Wang, B.; Nie, Z.; Lei, T.; Li, X.; et al. Routine pre-procedural rectal indometacin versus selective post-procedural rectal indometacin to prevent pancreatitis in patients undergoing endoscopic retrograde cholangiopancreatography: A multicentre, single-blinded, randomised controlled trial. Lancet Lond. Engl. 2016, 387, 2293–2301. [Google Scholar] [CrossRef]
- Zhu, C.; Fuchs, C.D.; Halilbasic, E.; Trauner, M. Bile acids in regulation of inflammation and immunity: Friend or foe? Clin. Exp. Rheumatol. 2016, 34, 25–31. [Google Scholar]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef]
- Jia, L.; Chen, H.; Yang, J.; Fang, X.; Niu, W.; Zhang, M.; Li, J.; Pan, X.; Ren, Z.; Sun, J.; et al. Combinatory antibiotic treatment protects against experimental acute pancreatitis by suppressing gut bacterial translocation to pancreas and inhibiting NLRP3 inflammasome pathway. Innate Immun. 2020, 26, 48–61. [Google Scholar] [CrossRef]


{kind=link}
| Compound | Type | Target | References |
|---|---|---|---|
| MCC950 | Diarylsulphonylurea | NLRP3 (ASC oligomerization) | [42] |
| Glyburide | Sulphonylurea | NLRP3 (ATP-sensitive K+ channels) | [108] |
| Emodin | Anthraquinone | Nrf2/ NF-κB/ NLRP3/ P2X7 | [109,110] |
| Danshensu | Phenolic acid | NF-κB/ STAT3/ NLRP3 | [111] |
| Fraxinellone | Limonoid | NLRP3 (CARD, caspase-1, IL1β, IL18) | [112] |
| Withaferin A | Alkaloid | NF-κB/ NLRP3 | [113] |
| Rutin | Flavonoid | NLRP3 (ASC, caspase-1) | [114] |
| Sulforaphane | Isothiocyanate | Nrf2/ NLRP3 | [115] |
| Cordycepin | Adenosine analogue | NF-κB/NLRP3 | [116] |
| Indomethacin | COX-2 inhibitor | NLRP3 (ASC, IL1β) | [117] |
| Iguratimod | COX-2 inhibitor | NF-κB/ NLRP3 | [118] |
| Apocynin | NOX inhibitor | NF-κB/ NLRP3 | [119] |
| INT-777 | Bile acid receptor agonist | ROS/ NLRP3 | [120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrero-Andrés, A.; Panisello-Roselló, A.; Roselló-Catafau, J.; Folch-Puy, E. NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis. Int. J. Mol. Sci. 2020, 21, 5386. https://doi.org/10.3390/ijms21155386
Ferrero-Andrés A, Panisello-Roselló A, Roselló-Catafau J, Folch-Puy E. NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis. International Journal of Molecular Sciences. 2020; 21(15):5386. https://doi.org/10.3390/ijms21155386
Chicago/Turabian StyleFerrero-Andrés, Ana, Arnau Panisello-Roselló, Joan Roselló-Catafau, and Emma Folch-Puy. 2020. "NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis" International Journal of Molecular Sciences 21, no. 15: 5386. https://doi.org/10.3390/ijms21155386
APA StyleFerrero-Andrés, A., Panisello-Roselló, A., Roselló-Catafau, J., & Folch-Puy, E. (2020). NLRP3 Inflammasome-Mediated Inflammation in Acute Pancreatitis. International Journal of Molecular Sciences, 21(15), 5386. https://doi.org/10.3390/ijms21155386