Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway



{kind=link}
Abstract
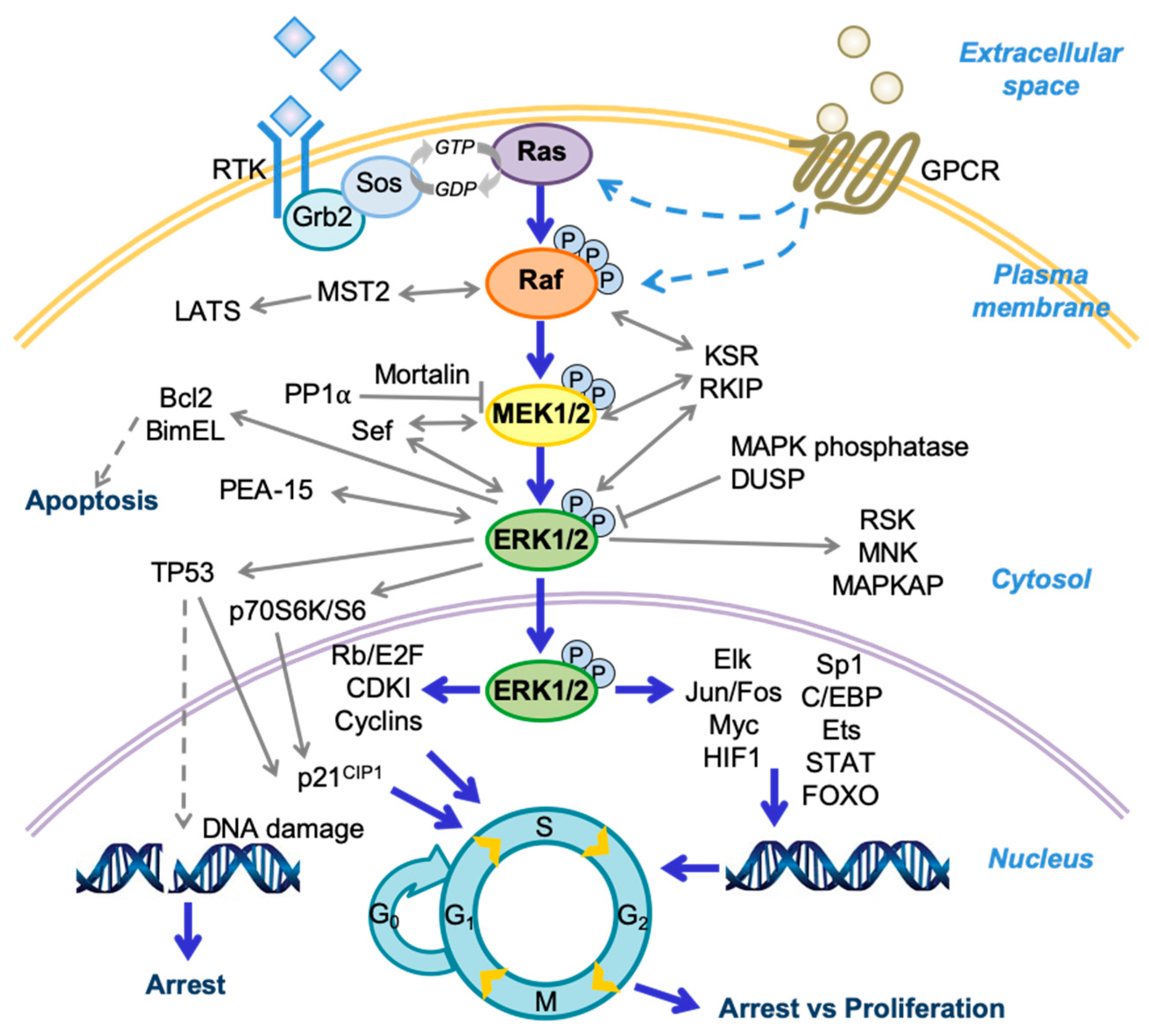
:1. Introduction
2. Mechanisms of Raf/MEK/ERK Growth Inhibitory Signaling
2.1. Non-Canonical Effects of Raf Are Also Involved in Growth Inhibitory Signaling
2.2. Differential Regulation of MEK1 and MEK2 Levels in Cells
2.3. Intrinsic Properties of ERK1/2 Affecting the Cell Fate Toward Growth Arrest Versus Death
2.4. Ectopic Expression of Autophosphorylating ERK2 Mutant Can Induce Cell Cycle Arrest
2.5. Regulators for Fine Tuning of Pathway Activity in Growth Inhibitory Signaling
3. Future Perspective
4. Conclusion
Funding
Acknowledgments
Conflicts of Interest
References
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Watson, U.; Vasudevan, L.; Saini, D.K. ERK Activation Pathways Downstream of GPCRs. Int. Rev. Cell Mol. Biol. 2018, 338, 79–109. [Google Scholar]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res. 2018, 138, 99–142. [Google Scholar] [PubMed]
- Unal, E.B.; Uhlitz, F.; Bluthgen, N. A compendium of ERK targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zheng, L.; Chng, W.J.; Ding, J.L. Comprehensive Analysis of ERK1/2 Substrates for Potential Combination Immunotherapies. Trends Pharmacol. Sci. 2019, 40, 897–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed] [Green Version]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Yaeger, R.; Corcoran, R.B. Targeting Alterations in the RAF-MEK Pathway. Cancer Discov. 2019, 9, 329–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Wu, P.K.; Park, J.I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Subramaniam, S.; Unsicker, K. ERK and cell death: ERK1/2 in neuronal death. FEBS J. 2010, 277, 22–29. [Google Scholar] [CrossRef]
- Park, J.I. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front. Biol. 2014, 9, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Thaler, S.; Hahnel, P.S.; Schad, A.; Dammann, R.; Schuler, M. RASSF1A mediates p21Cip1/Waf1-dependent cell cycle arrest and senescence through modulation of the Raf-MEK-ERK pathway and inhibition of Akt. Cancer Res. 2009, 69, 1748–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguria, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Zheng, X.; Huang, H.Y.; Shapiro, E.; Lepor, H.; Cordon-Cardo, C.; Sun, T.T.; Wu, X.R. Hyperactivation of Ha-ras oncogene, but not Ink4a/Arf deficiency, triggers bladder tumorigenesis. J. Clin. Invest. 2007, 117, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernandez-Porras, I.; Canamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence--halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [Green Version]
- McDuff, F.K.; Turner, S.D. Jailbreak: Oncogene-induced senescence and its evasion. Cell Signal. 2011, 23, 6–13. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Kamata, T.; Hussain, J.; Giblett, S.; Hayward, R.; Marais, R.; Pritchard, C. BRAF inactivation drives aneuploidy by deregulating CRAF. Cancer Res. 2010, 70, 8475–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, P.; Ambrogio, C.; Esteban-Burgos, L.; Gomez-Lopez, G.; Blasco, M.T.; Yao, Z.; Marais, R.; Rosen, N.; Chiarle, R.; Pisano, D.G.; et al. A Braf kinase-inactive mutant induces lung adenocarcinoma. Nature 2017, 548, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Romano, D.; Nguyen, L.K.; Matallanas, D.; Halasz, M.; Doherty, C.; Kholodenko, B.N.; Kolch, W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat. Cell Biol. 2014, 16, 673–684. [Google Scholar] [CrossRef]
- Rauch, J.; Vandamme, D.; Mack, B.; McCann, B.; Volinsky, N.; Blanco, A.; Gires, O.; Kolch, W. Differential localization of A-Raf regulates MST2-mediated apoptosis during epithelial differentiation. Cell Death Differ. 2016, 23, 1283–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, T.; Takano, K.; Yoshida, A.; Katada, F.; Sun, P.; Takenawa, T.; Andoh, T.; Endo, T. DA-Raf1, a competent intrinsic dominant-negative antagonist of the Ras-ERK pathway, is required for myogenic differentiation. J. Cell Biol. 2007, 177, 781–793. [Google Scholar] [CrossRef]
- Rauch, J.; Moran-Jones, K.; Albrecht, V.; Schwarzl, T.; Hunter, K.; Gires, O.; Kolch, W. c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res. 2011, 71, 4664–4674. [Google Scholar] [CrossRef] [Green Version]
- Kanno, E.; Kawasaki, O.; Takahashi, K.; Takano, K.; Endo, T. DA-Raf, a dominant-negative antagonist of the Ras-ERK pathway, is a putative tumor suppressor. Exp. Cell Res. 2018, 362, 111–120. [Google Scholar] [CrossRef]
- Ferrell, J.E., Jr. Tripping the switch fantastic: How a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 1996, 21, 460–466. [Google Scholar] [CrossRef]
- Hong, S.K.; Wu, P.K.; Karkhanis, M.; Park, J.I. ERK1/2 can feedback-regulate cellular MEK1/2 levels. Cell Signal. 2015, 27, 1939–1948. [Google Scholar] [CrossRef] [Green Version]
- Mansour, S.J.; Candia, J.M.; Matsuura, J.E.; Manning, M.C.; Ahn, N.G. Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry 1996, 35, 15529–15536. [Google Scholar] [CrossRef] [PubMed]
- Voisin, L.; Julien, C.; Duhamel, S.; Gopalbhai, K.; Claveau, I.; Saba-El-Leil, M.K.; Rodrigue-Gervais, I.G.; Gaboury, L.; Lamarre, D.; Basik, M.; et al. Activation of MEK1 or MEK2 isoform is sufficient to fully transform intestinal epithelial cells and induce the formation of metastatic tumors. BMC Cancer 2008, 8, 337. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.K.; Yoon, S.; Moelling, C.; Arthan, D.; Park, J.I. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. J. Biol. Chem. 2009, 284, 33006–33018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guegan, J.P.; Ezan, F.; Gailhouste, L.; Langouet, S.; Baffet, G. MEK1/2 overactivation can promote growth arrest by mediating ERK1/2-dependent phosphorylation of p70S6K. J. Cell Physiol. 2014, 229, 903–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Noh, S.J.; Zhou, G.; Dixon, J.E.; Guan, K.L. Selective activation of MEK1 but not MEK2 by A-Raf from epidermal growth factor-stimulated Hela cells. J. Biol. Chem. 1996, 271, 3265–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffer, H.J.; Catling, A.D.; Eblen, S.T.; Collier, L.S.; Krauss, A.; Weber, M.J. MP1: A MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 1998, 281, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Giroux, S.; Tremblay, M.; Bernard, D.; Cardin-Girard, J.F.; Aubry, S.; Larouche, L.; Rousseau, S.; Huot, J.; Landry, J.; Jeannotte, L.; et al. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 1999, 9, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Belanger, L.F.; Roy, S.; Tremblay, M.; Brott, B.; Steff, A.M.; Mourad, W.; Hugo, P.; Erikson, R.; Charron, J. Mek2 is dispensable for mouse growth and development. Mol. Cell Biol. 2003, 23, 4778–4787. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, V.; Guillemette, S.; Belanger, L.F.; Jacob, O.; Roy, S.; Charron, J. Map2k1 and Map2k2 genes contribute to the normal development of syncytiotrophoblasts during placentation. Development 2009, 136, 1363–1374. [Google Scholar] [CrossRef] [Green Version]
- Scholl, F.A.; Dumesic, P.A.; Barragan, D.I.; Harada, K.; Charron, J.; Khavari, P.A. Selective role for Mek1 but not Mek2 in the induction of epidermal neoplasia. Cancer Res. 2009, 69, 3772–3778. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.Y.; Rao, J.N.; Zou, T.; Liu, L.; Xiao, L.; Yu, T.X.; Turner, D.J.; Gorospe, M.; Wang, J.Y. Post-transcriptional regulation of MEK-1 by polyamines through the RNA-binding protein HuR modulating intestinal epithelial apoptosis. Biochem. J. 2010, 426, 293–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, W.B.; Kaji, K.; Kunath, T. ERK2 suppresses self-renewal capacity of embryonic stem cells, but is not required for multi-lineage commitment. PLoS ONE 2013, 8, e60907. [Google Scholar] [CrossRef] [Green Version]
- Saulnier, N.; Guihard, S.; Holy, X.; Decembre, E.; Jurdic, P.; Clay, D.; Feuillet, V.; Pages, G.; Pouyssegur, J.; Porteu, F.; et al. ERK1 regulates the hematopoietic stem cell niches. PLoS ONE 2012, 7, e30788. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.M.; Katayama, C.D.; Pages, G.; Pouyssegur, J.; Hedrick, S.M. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 2005, 23, 431–443. [Google Scholar] [CrossRef]
- Pages, G.; Guerin, S.; Grall, D.; Bonino, F.; Smith, A.; Anjuere, F.; Auberger, P.; Pouyssegur, J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999, 286, 1374–1377. [Google Scholar]
- Busca, R.; Christen, R.; Lovern, M.; Clifford, A.M.; Yue, J.X.; Goss, G.G.; Pouyssegur, J.; Lenormand, P. ERK1 and ERK2 present functional redundancy in tetrapods despite higher evolution rate of ERK1. BMC Evol. Biol. 2015, 15, 179. [Google Scholar] [CrossRef] [Green Version]
- Lefloch, R.; Pouyssegur, J.; Lenormand, P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell Biol. 2008, 28, 511–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.K.; Wu, P.K.; Park, J.I. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell Signal. 2018, 42, 11–20. [Google Scholar] [CrossRef]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef] [Green Version]
- Leung, G.P.; Feng, T.; Sigoillot, F.D.; Geyer, F.C.; Shirley, M.D.; Ruddy, D.A.; Rakiec, D.P.; Freeman, A.K.; Engelman, J.A.; Jaskelioff, M.; et al. Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-mutant Melanoma. Mol. Cancer Res. 2019, 17, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Whelan, J.T.; Hollis, S.E.; Cha, D.S.; Asch, A.S.; Lee, M.H. Post-transcriptional regulation of the Ras-ERK/MAPK signaling pathway. J. Cell Physiol. 2012, 227, 1235–1241. [Google Scholar] [CrossRef]
- Lee, M.H.; Hook, B.; Pan, G.; Kershner, A.M.; Merritt, C.; Seydoux, G.; Thomson, J.A.; Wickens, M.; Kimble, J. Conserved regulation of MAP kinase expression by PUF RNA-binding proteins. PLoS Genet. 2007, 3, e233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, P.S.; Whalen, A.M.; Tolwinski, N.S.; Wilsbacher, J.; Froelich-Ammon, S.J.; Garcia, M.; Osheroff, N.; Ahn, N.G. Extracellular signal-regulated kinase activates topoisomerase IIalpha through a mechanism independent of phosphorylation. Mol. Cell Biol. 1999, 19, 3551–3560. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Crespo, P. Working without kinase activity: Phosphotransfer-independent functions of extracellular signal-regulated kinases. Sci. Signal. 2011, 4, re3. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Gotoh, Y.; Nishida, E. Interaction of MAP kinase with MAP kinase kinase: Its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J. 1997, 16, 1901–1908. [Google Scholar] [CrossRef] [Green Version]
- Vantaggiato, C.; Formentini, I.; Bondanza, A.; Bonini, C.; Naldini, L.; Brambilla, R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J. Biol. 2006, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Barr, D.; Oashi, T.; Burkhard, K.; Lucius, S.; Samadani, R.; Zhang, J.; Shapiro, P.; MacKerell, A.D.; van der Vaart, A. Importance of domain closure for the autoactivation of ERK2. Biochemistry 2011, 50, 8038–8048. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Strand, A.; Robbins, D.; Cobb, M.H.; Goldsmith, E.J. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature 1994, 367, 704–711. [Google Scholar] [CrossRef]
- Robbins, D.J.; Zhen, E.; Owaki, H.; Vanderbilt, C.A.; Ebert, D.; Geppert, T.D.; Cobb, M.H. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J. Biol. Chem. 1993, 268, 5097–5106. [Google Scholar]
- Rossomando, A.J.; Wu, J.; Michel, H.; Shabanowitz, J.; Hunt, D.F.; Weber, M.J.; Sturgill, T.W. Identification of Tyr-185 as the site of tyrosine autophosphorylation of recombinant mitogen-activated protein kinase p42mapk. Proc. Natl. Acad. Sci. USA 1992, 89, 5779–5783. [Google Scholar] [CrossRef] [Green Version]
- Seger, R.; Ahn, N.G.; Boulton, T.G.; Yancopoulos, G.D.; Panayotatos, N.; Radziejewska, E.; Ericsson, L.; Bratlien, R.L.; Cobb, M.H.; Krebs, E.G. Microtubule-associated protein 2 kinases, ERK1 and ERK2, undergo autophosphorylation on both tyrosine and threonine residues: Implications for their mechanism of activation. Proc. Natl. Acad. Sci. USA 1991, 88, 6142–6146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emrick, M.A.; Hoofnagle, A.N.; Miller, A.S.; Ten Eyck, L.F.; Ahn, N.G. Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J. Biol. Chem. 2001, 276, 46469–46479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emrick, M.A.; Lee, T.; Starkey, P.J.; Mumby, M.C.; Resing, K.A.; Ahn, N.G. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc. Natl. Acad. Sci. USA 2006, 103, 18101–18106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.K.; Hong, S.K.; Yoon, S.H.; Park, J.I. Active ERK2 is sufficient to mediate growth arrest and differentiation signaling. FEBS J. 2015, 282, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Warthaka, M.; Yan, C.; Kaoud, T.S.; Ren, P.; Dalby, K.N. Examining docking interactions on ERK2 with modular peptide substrates. Biochemistry 2011, 50, 9500–9510. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef]
- Murphy, L.O.; MacKeigan, J.P.; Blenis, J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol. Cell Biol. 2004, 24, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.; Hadfield, K.; Howes, E.; Cook, S.J. Identification of a DEF-type docking domain for extracellular signal-regulated kinases 1/2 that directs phosphorylation and turnover of the BH3-only protein BimEL. J. Biol. Chem. 2005, 280, 17657–17663. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.D.; Sacks, D.B. Protein scaffolds in MAP kinase signalling. Cell Signal. 2009, 21, 462–469. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.M.; Keyse, S.M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007, 26, 3203–3213. [Google Scholar] [CrossRef] [Green Version]
- Callaway, K.; Abramczyk, O.; Martin, L.; Dalby, K.N. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry 2007, 46, 9187–9198. [Google Scholar] [PubMed]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Gaumont-Leclerc, M.F.; Mukhopadhyay, U.K.; Goumard, S.; Ferbeyre, G. PEA-15 is inhibited by adenovirus E1A and plays a role in ERK nuclear export and Ras-induced senescence. J. Biol. Chem. 2004, 279, 46802–46809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomeusz, C.; Rosen, D.; Wei, C.; Kazansky, A.; Yamasaki, F.; Takahashi, T.; Itamochi, H.; Kondo, S.; Liu, J.; Ueno, N.T. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer Res. 2008, 68, 9302–9310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrak, S.C.; Renault-Mihara, F.; Parraga, M.; Bogerd, J.; van de Kant, H.J.; Lopez-Casas, P.P.; Paz, M.; del Mazo, J.; de Rooij, D.G. Phosphoprotein enriched in astrocytes-15 is expressed in mouse testis and protects spermatocytes from apoptosis. Reproduction 2007, 133, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Furthauer, M.; Lin, W.; Ang, S.L.; Thisse, B.; Thisse, C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat. Cell Biol. 2002, 4, 170–174. [Google Scholar] [CrossRef]
- Tsang, M.; Friesel, R.; Kudoh, T.; Dawid, I.B. Identification of Sef, a novel modulator of FGF signalling. Nat. Cell Biol. 2002, 4, 165–169. [Google Scholar] [CrossRef]
- Preger, E.; Ziv, I.; Shabtay, A.; Sher, I.; Tsang, M.; Dawid, I.B.; Altuvia, Y.; Ron, D. Alternative splicing generates an isoform of the human Sef gene with altered subcellular localization and specificity. Proc. Natl. Acad. Sci. USA 2004, 101, 1229–1234. [Google Scholar] [CrossRef] [Green Version]
- Conlon, P.; Gelin-Licht, R.; Ganesan, A.; Zhang, J.; Levchenko, A. Single-cell dynamics and variability of MAPK activity in a yeast differentiation pathway. Proc. Natl. Acad. Sci. USA 2016, 113, E5896–E5905. [Google Scholar] [CrossRef] [Green Version]
- Kortum, R.L.; Johnson, H.J.; Costanzo, D.L.; Volle, D.J.; Razidlo, G.L.; Fusello, A.M.; Shaw, A.S.; Lewis, R.E. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol. Cell Biol. 2006, 26, 2202–2214. [Google Scholar] [CrossRef] [Green Version]
- Razidlo, G.L.; Johnson, H.J.; Stoeger, S.M.; Cowan, K.H.; Bessho, T.; Lewis, R.E. KSR1 is required for cell cycle reinitiation following DNA damage. J. Biol. Chem. 2009, 284, 6705–6715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuierer, M.M.; Bataille, F.; Hagan, S.; Kolch, W.; Bosserhoff, A.K. Reduction in Raf kinase inhibitor protein expression is associated with increased Ras-extracellular signal-regulated kinase signaling in melanoma cell lines. Cancer Res. 2004, 64, 5186–5192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrell Stewart, D.R.; Clark, G.J. Pumping the brakes on RAS - negative regulators and death effectors of RAS. J. Cell Sci. 2020, 133, jcs238865. [Google Scholar] [CrossRef]
- Wu, P.K.; Hong, S.K.; Veeranki, S.; Karkhanis, M.; Starenki, D.; Plaza, J.A.; Park, J.I. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol. Cell Biol. 2013, 33, 4051–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.K.; Hong, S.K.; Park, J.I. Steady-State Levels of Phosphorylated Mitogen-Activated Protein Kinase Kinase 1/2 Determined by Mortalin/HSPA9 and Protein Phosphatase 1 Alpha in KRAS and BRAF Tumor Cells. Mol. Cell Biol. 2017, 37, e00061-17. [Google Scholar] [CrossRef] [Green Version]
- Karkhanis, M.; Park, J.I. Sp1 regulates Raf/MEK/ERK-induced p21(CIP1) transcription in TP53-mutated cancer cells. Cell Signal. 2015, 27, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Deschenes-Simard, X.; Gaumont-Leclerc, M.F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013, 27, 900–915. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.K.; Hong, S.K.; Chen, W.; Becker, A.E.; Gundry, R.L.; Lin, C.W.; Shao, H.; Gestwicki, J.E.; Park, J.I. Mortalin (HSPA9) facilitates BRAF-mutant tumor cell survival by suppressing ANT3-mediated mitochondrial membrane permeability. Sci. Signal. 2020, 13, eaay1478. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.K.; Hong, S.K.; Starenki, D.; Oshima, K.; Shao, H.; Gestwicki, J.E.; Tsai, S.; Park, J.I. Mortalin/HSPA9 targeting selectively induces KRAS tumor cell death by perturbing mitochondrial membrane permeability. Oncogene 2020, 39, 4257–4270. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 2015, 34, 4624–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starenki, D.; Park, J.I. Selective Mitochondrial Uptake of MKT-077 Can Suppress Medullary Thyroid Carcinoma Cell Survival In Vitro and In Vivo. Endocrinol Metab 2015, 30, 593–603. [Google Scholar] [CrossRef]
- Starenki, D.; Sosonkina, N.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.Y.; Niessen, H.W.M.; Rozeman, E.A.; Geukes Foppen, M.H.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Maddodi, N.; Huang, W.; Havighurst, T.; Kim, K.; Longley, B.J.; Setaluri, V. Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J. Invest. Dermatol. 2010, 130, 1657–1667. [Google Scholar] [CrossRef] [Green Version]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife 2018, 7, e33718. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, P.-K.; Becker, A.; Park, J.-I. Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. Int. J. Mol. Sci. 2020, 21, 5436. https://doi.org/10.3390/ijms21155436
Wu P-K, Becker A, Park J-I. Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. International Journal of Molecular Sciences. 2020; 21(15):5436. https://doi.org/10.3390/ijms21155436
Chicago/Turabian StyleWu, Pui-Kei, Andrew Becker, and Jong-In Park. 2020. "Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway" International Journal of Molecular Sciences 21, no. 15: 5436. https://doi.org/10.3390/ijms21155436
APA StyleWu, P. -K., Becker, A., & Park, J. -I. (2020). Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. International Journal of Molecular Sciences, 21(15), 5436. https://doi.org/10.3390/ijms21155436