The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
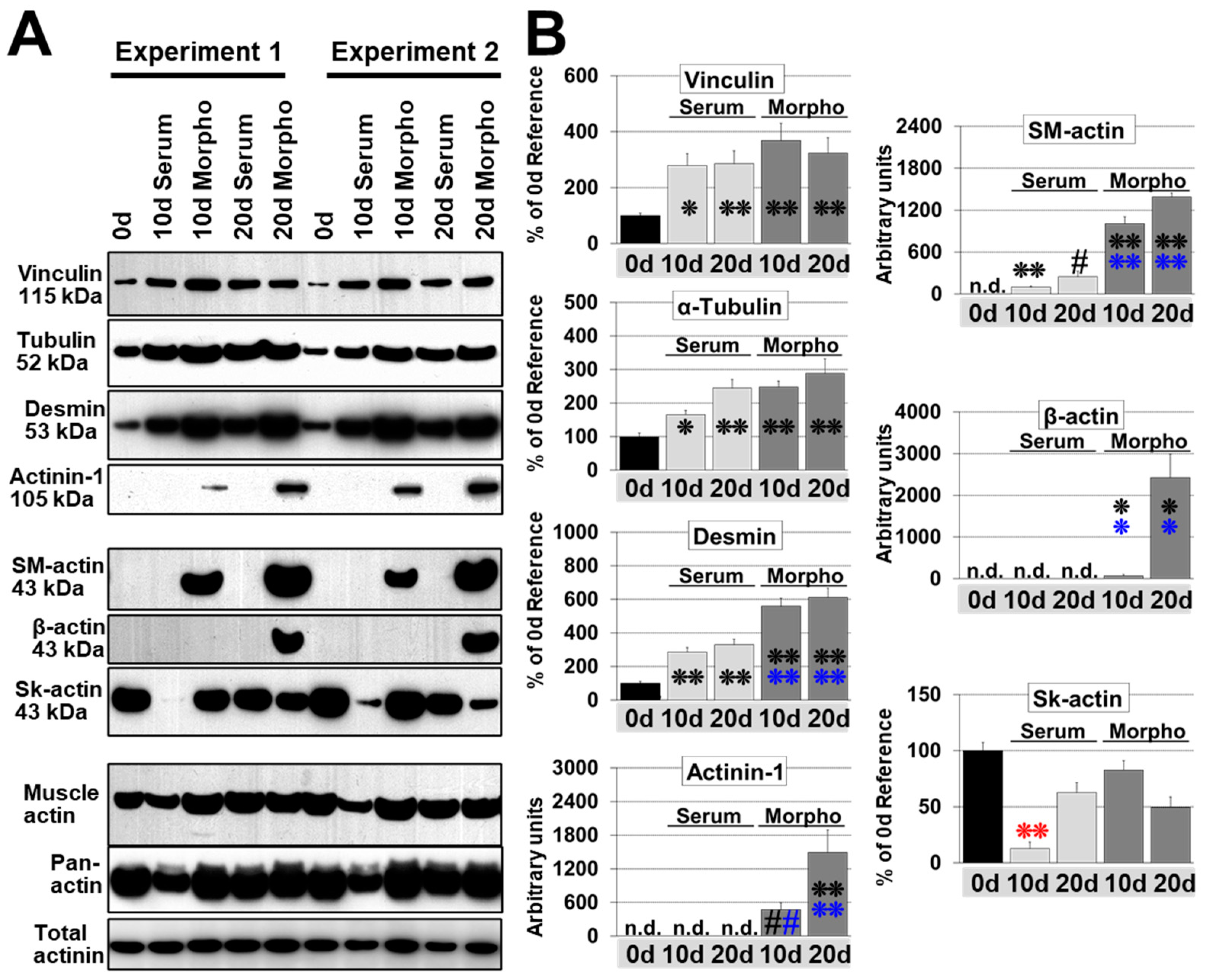
2.1. Reexpression of Fetal Genes, Cellular Morphology, and Atrophy Distinguish Fetal-Reprogrammed from Hypertrophic Cardiomyocytes
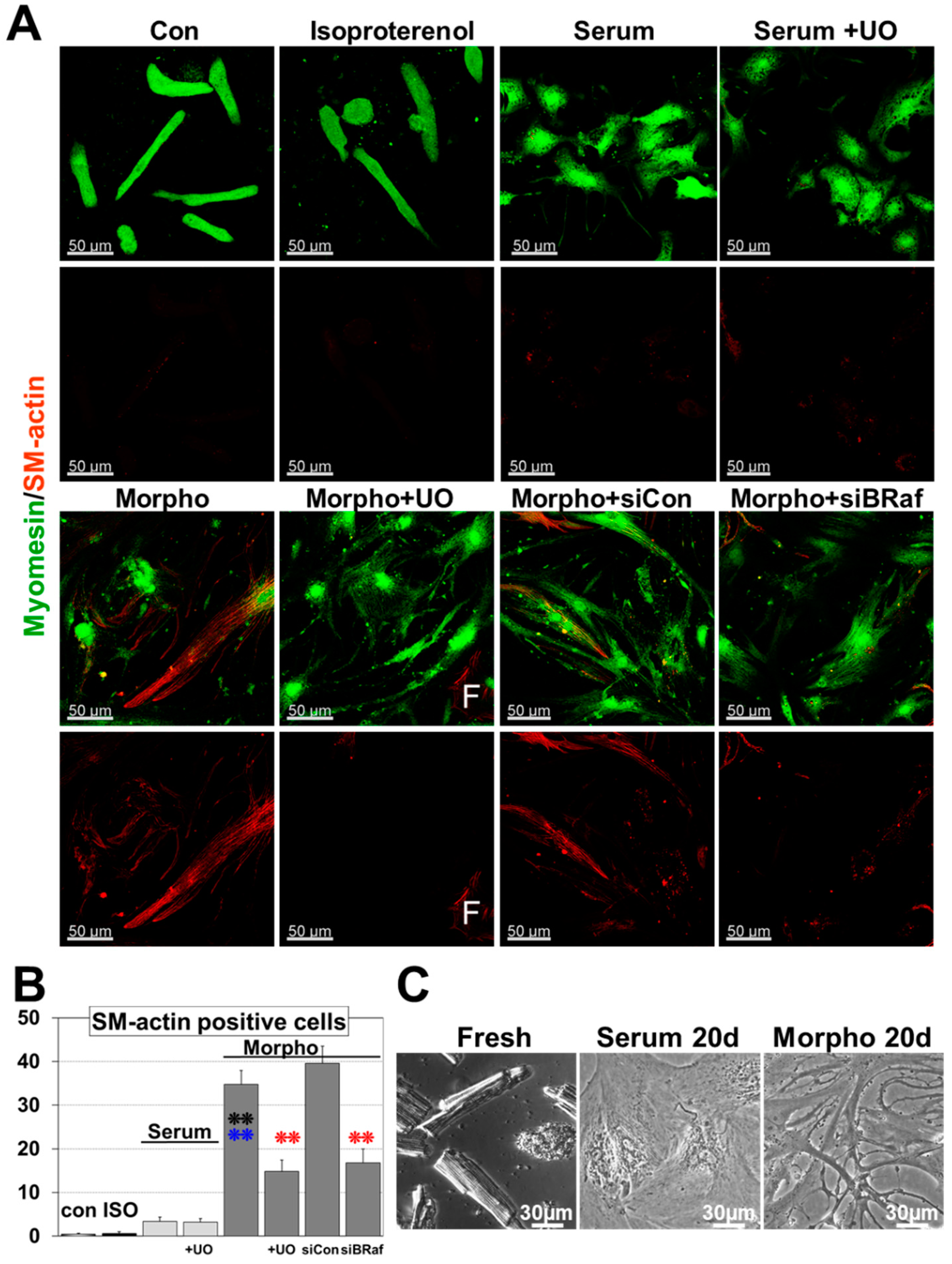
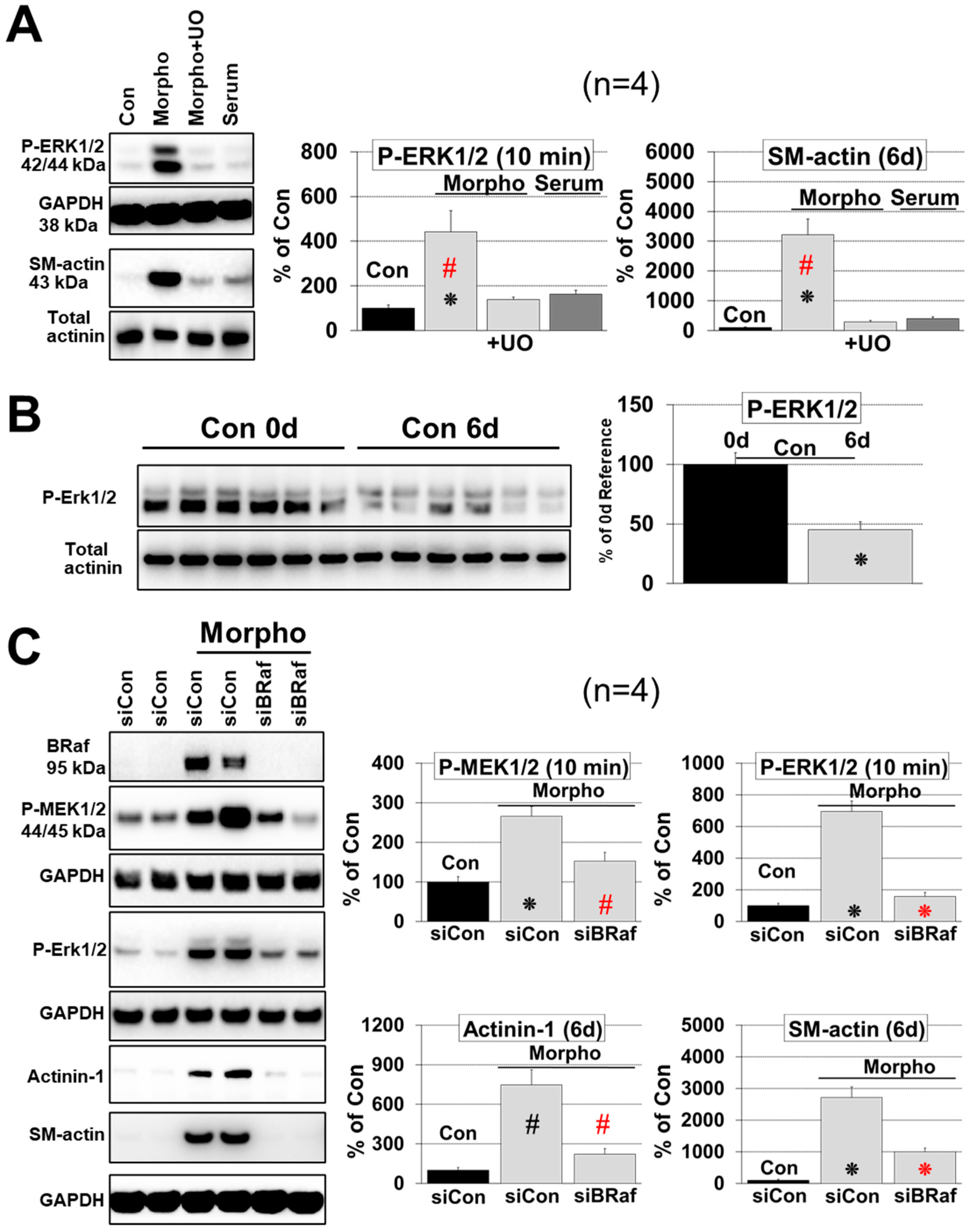
2.2. Morphogens Induce MAPK Dependent Ischemic Resistance and Fetal Remodeling
2.3. B-Raf Is Responsible for MAPK Mediated Fetal Remodeling and Ischemic Resistance
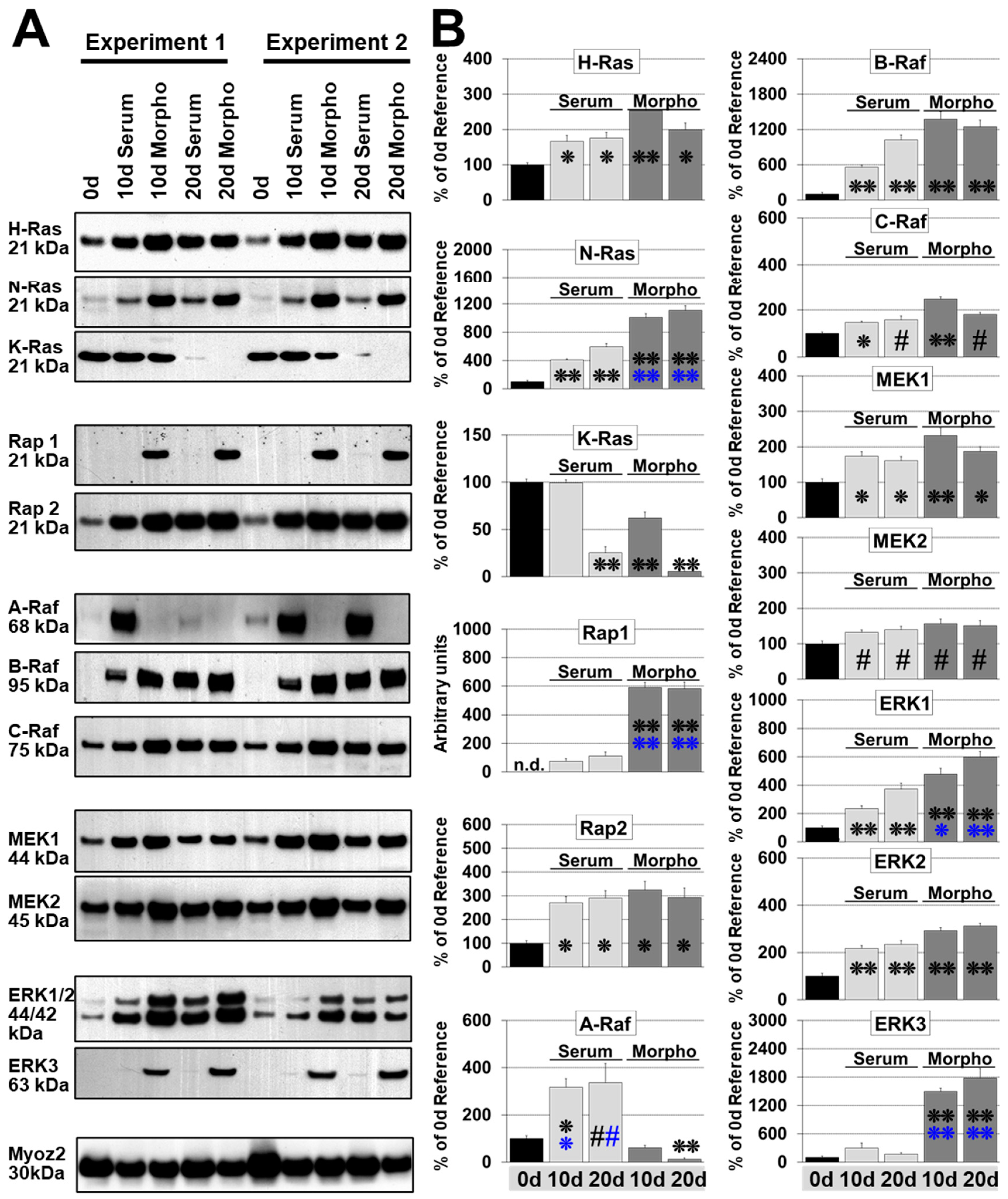
2.4. N-Ras Replaces K-Ras while H-Ras Increases Slightly during Remodeling
2.5. Rap1 Is Reexpressed during Fetal Remodeling
2.6. B-Raf and C-Raf Increase in Both Model Systems while A-Raf Increases Sharply During Hypertrophic Remodeling
2.7. MEK1/2 Increased Slightly and ERK1/2 Markedly in Both Models, but ERK3 Was Only Reexpressed during Fetal Remodeling
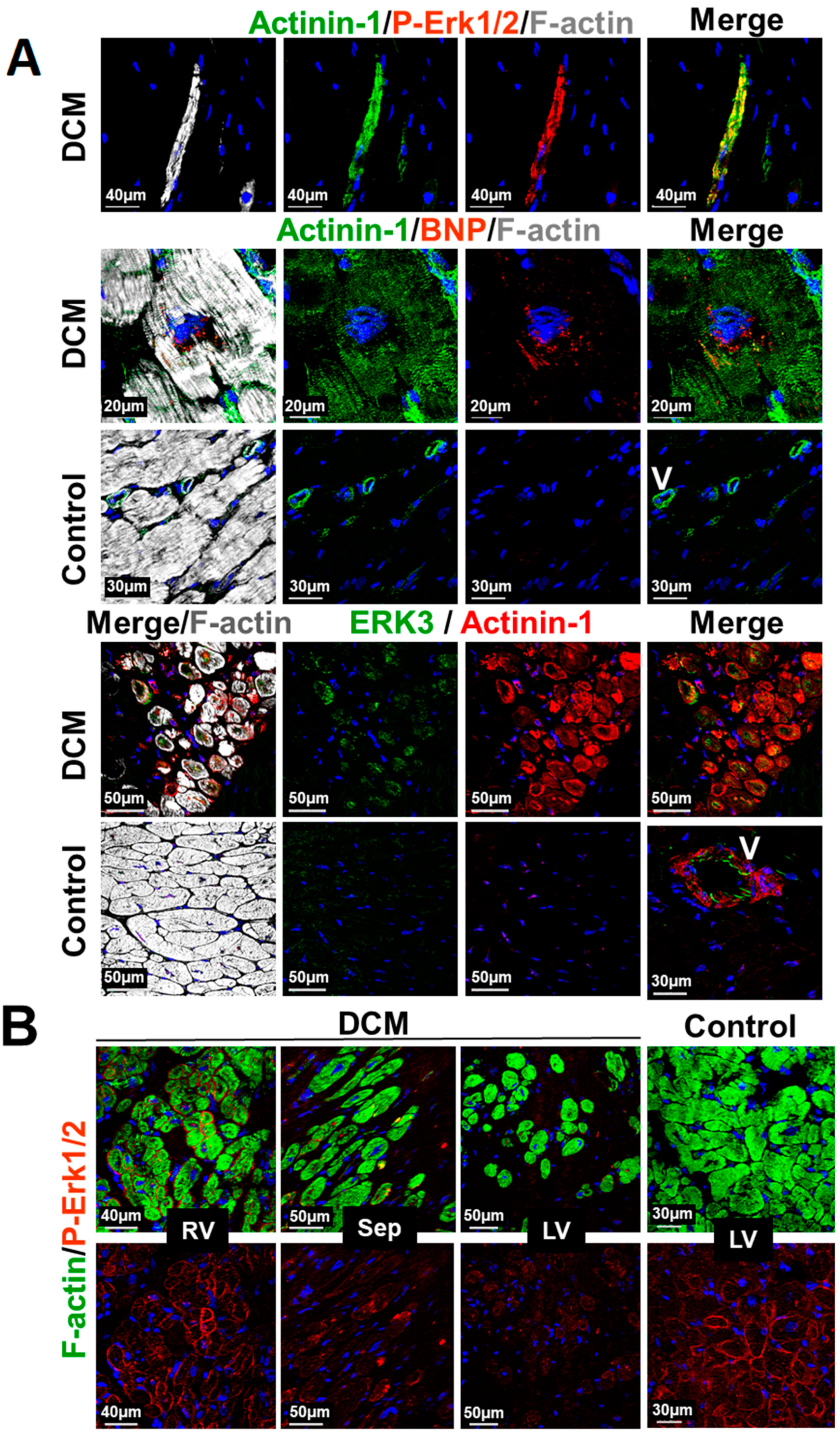
2.8. Structural and MAPK Remodeling Are Observed in Cardiomyocytes of Patients with Dilated Cardiomyopathy
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Settings
- Control (Con 0 d), day of stimulation start;
- Myocytes stimulated for 10 days with 5% serum (Serum);
- Myocytes stimulated for 10 days with endothelial morphogens (Morpho);
- Myocytes stimulated for 20 days with 5% serum (Serum);
- Myocytes stimulated for 20 days with endothelial morphogens (Morpho);
- In some experiments, we use a new control group of cultured cardiomyocytes in the absence of any stimulants for 6 days (unloaded; Con 6 d);
- To determine ischemic resistance, cultures were pretreated for 6 days and kept for 9 h at 1% O2 in glucose-free PBS;
- To determine MAPK-dependent fetal remodeling and cell size parameters, cultures were treated either for 10 min or 6 days;
- In order to determine the effect of B-Raf on MAPK activation and fetal remodeling, cardiomyocytes were pretreated for 3 days with control or B-Raf siRNA and then stimulated with morphogens.
4.2. Culture of Adult Cardiomyocytes, Basic Medium, and Preparation of Endothelial Morphogens
4.3. Determination of the Protein Content, Cell Size Parameters, and siRNA Knock-Down
4.4. Western Blot Analysis and Microscopy
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Approval
References
- Swynghedauw, B. Molecular mechanisms of myocardial remodeling. Am. J. Physiol. 1999, 79, 215–262. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac Remodeling—Concepts and Clinical Implications: A Consensus Paper From an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schonburg, M.; Elsasser, A.; Polyakova, V.; Bauer, E.P.; Klovekorn, W.-P.; Schaper, J. Progression From Compensated Hypertrophy to Failure in the Pressure-Overloaded Human Heart: Structural Deterioration and Compensatory Mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [Green Version]
- Hoshijima, M.; Chien, K.R. Mixed signals in heart failure. J. Clin. Investig. 2002, 109, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.; Crowell, K.; Proctor, A.; Jensen, B.C. Cardiovascular Effects of the MEK Inhibitor, Trametinib: A Case Report, Literature Review, and Consideration of Mechanism. Cardiovasc. Toxicol. 2017, 17, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Chen, H. Cardiotoxicity of Anticancer Therapeutics. Front. Cardiovasc. Med. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Force, T.; Krause, D.S.; Van Etten, R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 2007, 7, 332–344. [Google Scholar] [CrossRef]
- Bueno, O.F.; Molkentin, J.D. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ. Res. 2002, 91, 776–781. [Google Scholar] [CrossRef]
- Eitenmüller, I.; Volger, O.; Kluge, A.; Troidl, K.; Barancik, M.; Cai, W.J.; Heil, M.; Pipp, F.; Fischer, S.; Horrevoets, A.J.; et al. The range of adaptation by collateral vessels after femoral artery occlusion. Circ. Res. 2006, 99, 656–662. [Google Scholar] [CrossRef]
- Hunter, J.J.; Tanaka, N.; Rockman, H.A.; Ross Jr, J.; Chien, K.R. Ventricular expression of a MLC-2-v-ras fusion gene induces cardiac hypertrophy and selective diastolic dysfunction in transgenic mice. J. Biol. Chem. 1995, 270, 23173–23178. [Google Scholar] [CrossRef] [Green Version]
- Kubin, T.; Pöling, J.; Kostin, S.; Gajawada, P.; Hein, S.; Rees, W.; Wietelmann, A.; Tanaka, M.; Lörchner, H.; Schimanski, S.; et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell 2011, 9, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lörchner, H.; Pöling, J.; Gajawada, P.; Hou, Y.; Polyakova, V.; Kostin, S.; Adrian-Segarra, J.M.; Boettger, T.; Wietelmann, A.; Warnecke, H.; et al. Myocardial healing requires Reg3β-dependent accumulation of macrophages in the ischemic heart. Nat. Med. 2015, 21, 353–362. [Google Scholar] [CrossRef]
- Pöling, J.; Gajawada, P.; Richter, M.; Lörchner, H.; Polyakova, V.; Kostin, S.; Shin, J.; Boettger, T.; Walther, T.; Rees, W.; et al. Therapeutic targeting of the oncostatin M receptor-β prevents inflammatory heart failure. Basic Res. Cardiol. 2014, 109, 396. [Google Scholar] [CrossRef] [PubMed]
- Pöling, J.; Szibor, M.; Schimanski, S.; Ingelmann, M.E.; Rees, W.; Gajawada, P.; Kochfar, Z.; Lörchner, H.; Salwig, I.; Shin, J.Y.; et al. Induction of smooth muscle cell migration during arteriogenesis is mediated by Rap2. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2297–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.; Watanabe, S.; Hoelper, S.; Krüger, M.; Kostin, S.; Pöling, J.; Kubin, T.; Braun, T. BRAF activates PAX3 to control muscle precursor cell migration during forelimb muscle development. eLife 2016, 5, 3767. [Google Scholar] [CrossRef] [PubMed]
- Szibor, M.; Pöling, J.; Warnecke, H.; Kubin, T.; Braun, T. Remodeling and dedifferentiation of adult cardiomyocytes during disease and regeneration. Cell. Mol. Life Sci. 2014, 71, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Kubin, T.; von der Ahe, D.; Deindl, E.; Schaper, W.; Zimmermann, R. MEK hyperphosphorylation coincides with cell cycle shut down of cultured smooth muscle cells. J. Cell. Physiol. 2006, 206, 25–34. [Google Scholar] [CrossRef]
- Karoulia, Z.; Gavathiotis, E.; Poulikakos, P.I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 2017, 17, 676–691. [Google Scholar] [CrossRef]
- Wu, P.-K.; Park, J.-I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Delaney, A.M.; Printen, J.A.; Chen, H.; Fauman, E.B.; Dudley, D.T. Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol. Cell. Biol. 2002, 22, 7593–7602. [Google Scholar] [CrossRef] [Green Version]
- Kubin, T.; Cetinkaya, A.; Schönburg, M.; Beiras-Fernandez, A.; Walther, T.; Richter, M. The MEK1 inhibitors UO126 and PD98059 block PDGF-AB induced phosphorylation of threonine 292 in porcine smooth muscle cells. Cytokine 2017, 95, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Heling, A.; Zimmermann, R.; Kostin, S.; Maeno, Y.; Hein, S.; Devaux, B.; Bauer, E.; Klövekorn, W.-P.; Schlepper, M.; Schaper, W.; et al. Increased Expression of Cytoskeletal, Linkage, and Extracellular Proteins in Failing Human Myocardium. Circ. Res. 2000, 86, 846–853. [Google Scholar] [CrossRef] [Green Version]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Lautze, H.J.; Walther, T.; Braun, T.; Kostin, S.; Kubin, T. The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J. Heart Lung Transplant. 2015, 34, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, A.; Berge, B.; Sen-Hild, B.; Troidl, K.; Gajawada, P.; Kubin, N.; Valeske, K.; Schranz, D.; Akintürk, H.; Schönburg, M.; et al. Radixin Relocalization and Nonmuscle α-Actinin Expression Are Features of Remodeling Cardiomyocytes in Adult Patients with Dilated Cardiomyopathy. Dis. Markers 2020, 2020, 9356738. [Google Scholar] [CrossRef]
- Hein, S.; Block, T.; Zimmermann, R.; Kostin, S.; Scheffold, T.; Kubin, T.; Klövekorn, W.P.; Schaper, J. Deposition of nonsarcomeric alpha-actinin in cardiomyocytes from patients with dilated cardiomyopathy or chronic pressure overload. Exp. Clin. Cardiol. 2009, 14, e68–e75. [Google Scholar]
- Hou, Y.; Adrian-Segarra, J.M.; Richter, M.; Kubin, N.; Shin, J.; Werner, I.; Walther, T.; Schönburg, M.; Pöling, J.; Warnecke, H.; et al. Animal Models and “Omics” Technologies for Identification of Novel Biomarkers and Drug Targets to Prevent Heart Failure. BioMed Res. Int. 2015, 2015, 212910. [Google Scholar] [CrossRef]
- Kubin, T.; Ando, H.; Scholz, D.; Bramlage, P.; Kostin, S.; van Veen, A.; Hein, S.; Fischer, S.; Breier, A.; Schaper, J.; et al. Microvascular endothelial cells remodel cultured adult cardiomyocytes and increase survival. Am. J. Physiol. 1999, 45, H2179–H2187. [Google Scholar] [CrossRef]
- Mercer, K.; Chiloeches, A.; Hüser, M.; Kiernan, M.; Marais, R.; Pritchard, C. ERK signalling and oncogene transformation are not impaired in cells lacking A-Raf. Oncogene 2002, 21, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Stork, P.J.S. Does Rap1 deserve a bad Rap? Trends Biochem. Sci. 2003, 28, 267–275. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Kolch, W. Untying the regulation of the Raf-1 kinase. Arch. Biochem. Biophys. 2002, 404, 3–9. [Google Scholar] [CrossRef]
- Johnson, L.; Greenbaum, D.; Cichowski, K.; Mercer, K.; Murphy, E.; Schmitt, E.; Bronson, R.T.; Umanoff, H.; Edelmann, W.; Kucherlapati, R.; et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997, 11, 2468–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koera, K.; Nakamura, K.; Nakao, K.; Miyoshi, J.; Toyoshima, K.; Hatta, T.; Otani, H.; Aiba, A.; Katsuki, M. K-Ras is essential for the development of the mouse embryo. Oncogene 1997, 15, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, A.; Ehrhardt, G.R.A.; Guo, X.; Schrader, J.W. Ras and relatives-job sharing and networking keep an old family together. Exp. Hematol. 2002, 30, 1089–1106. [Google Scholar] [CrossRef]
- Chrzanowska-Wodnicka, M.; White, G.C., II; Quilliam, L.A.; Whitehead, K.J. Small GTPase Rap1 Is Essential for Mouse Development and Formation of Functional Vasculature. PLoS ONE 2015, 10, e0145689. [Google Scholar] [CrossRef] [Green Version]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Revs. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Barnier, J.V.; Papin, C.; Eychene, A.; Lecoq, O.; Calothy, G. The Mouse B-raf Encodes Multiple Protein Isoforms with Tissue-specific Expression. J. Biol. Chem. 1995, 270, 23381–23389. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.O.; Irwin, P.; Katz, S.; Pelech, S.L. Expression of Mitogen-Activated Protein Kinase Pathways During Postnatal Development of Rat Heart. J. Cell. Biochem. 1998, 71, 286–301. [Google Scholar] [CrossRef]
- Luckett, J.C.A.; Hüser, M.B.; Giagtzoglou, N.; Brown, J.E.; Pritchard, C.A. Expression of the A-raf Proto-Oncogene in the Normal Adult and Embryonic Mouse. Cell Growth Differ. 2000, 11, 163–171. [Google Scholar]
- Bogoyevitch, M.A.; Marshall, C.J.; Sugden, P.H. Hypertrophic agonists stimulate the activities of the protein kinases c-Raf and A-Raf in cultured ventricular myocytes. J. Biol. Chem. 1995, 270, 26303–26310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, H.; Vikis, H.G.; Guan, K.L. Mechanisms of regulating the Raf kinase family. Cell. Signal. 2003, 15, 463–469. [Google Scholar] [CrossRef]
- Mercer, K.; Pritchard, C.A. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 2003, 1653, 25–40. [Google Scholar] [CrossRef]
- Hagemann, C.; Rapp, U. Isotype-Specific Functions of Raf Kinases. Exp. Cell. Res. 1999, 253, 34–46. [Google Scholar] [CrossRef]
- van Bilsen, M.; van der Tusse, G.J.; Reneman, R.S. Transcriptional regulation of metabolic processes: Implications for cardiac metabolism. Eur. J. Physiol. 1998, 437, 2–14. [Google Scholar] [CrossRef]
- Elkhadragy, L.; Alsaran, H.; Long, W. The C-Terminus Tail Regulates ERK3 Kinase Activity and Its Ability in Promoting Cancer Cell Migration and Invasion. Int. J. Mol. Sci. 2020, 21, 4044. [Google Scholar] [CrossRef]
- Klinger, S.; Turgeon, B.; Lévesque, K.; Wood, G.A.; Aagaard-Tillery, K.M.; Meloche, S. Loss of Erk3 function in mice leads to intrauterine growth restriction, pulmonary immaturity, and neonatal lethality. Proc. Natl. Acad. Sci. USA 2009, 106, 16710–16715. [Google Scholar] [CrossRef] [Green Version]
- Piper, H.M. Adult ventricular rat heart muscle cells. In CellCulture Techniques in Heart and Vessel Research; Springer: Berlin/Heidelberg, Germany, 1990; pp. 36–60. [Google Scholar]
- Ando, H.; Kubin, T.; Schaper, W.; Schaper, J. Porcine coronary microvascular endothelial cells express a-smooth muscle actin and show low NOS III activity. Am. J. Physiol. 1999, 45, H1755–H1768. [Google Scholar]
- West, D.C.; Sattar, A.; Kumar, S. A simplified in situ solubilization procedure for the determination of DNA and cell number in tissue cultured mammalian cells. Anal. Biochem. 1985, 147, 289–295. [Google Scholar] [CrossRef]
- Kubin, T.; Tomars, M.; Fach, C.; Hein, S.; Bramlage, P.; Shim, G.J.; Scholz, D.; Kostin, S.; Zimmermann, R.; Elsässer, A.; et al. Transforming growth factor-beta1 downregulates beating frequency and remodeling of cultured rat adult cardiomyocytes. Cell Tissue Res. 2005, 321, 57–66. [Google Scholar] [CrossRef]
- Kubin, N.; Richter, M.; Sen-Hild, B.; Akintürk, H.; Schönburg, M.; Kubin, T.; Cetinkaya, A. Macrophages represent the major pool of IL-7Rα expressing cells in patients with myocarditis. Cytokine 2020, 130, 155053. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubin, T.; Cetinkaya, A.; Kubin, N.; Bramlage, P.; Sen-Hild, B.; Gajawada, P.; Akintürk, H.; Schönburg, M.; Schaper, W.; Choi, Y.-H.; et al. The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes. Int. J. Mol. Sci. 2020, 21, 6348. https://doi.org/10.3390/ijms21176348
Kubin T, Cetinkaya A, Kubin N, Bramlage P, Sen-Hild B, Gajawada P, Akintürk H, Schönburg M, Schaper W, Choi Y-H, et al. The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes. International Journal of Molecular Sciences. 2020; 21(17):6348. https://doi.org/10.3390/ijms21176348
Chicago/Turabian StyleKubin, Thomas, Ayse Cetinkaya, Natalia Kubin, Peter Bramlage, Bedriye Sen-Hild, Praveen Gajawada, Hakan Akintürk, Markus Schönburg, Wolfgang Schaper, Yeong-Hoon Choi, and et al. 2020. "The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes" International Journal of Molecular Sciences 21, no. 17: 6348. https://doi.org/10.3390/ijms21176348
APA StyleKubin, T., Cetinkaya, A., Kubin, N., Bramlage, P., Sen-Hild, B., Gajawada, P., Akintürk, H., Schönburg, M., Schaper, W., Choi, Y. -H., Barancik, M., & Richter, M. (2020). The MEK/ERK Module Is Reprogrammed in Remodeling Adult Cardiomyocytes. International Journal of Molecular Sciences, 21(17), 6348. https://doi.org/10.3390/ijms21176348