Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
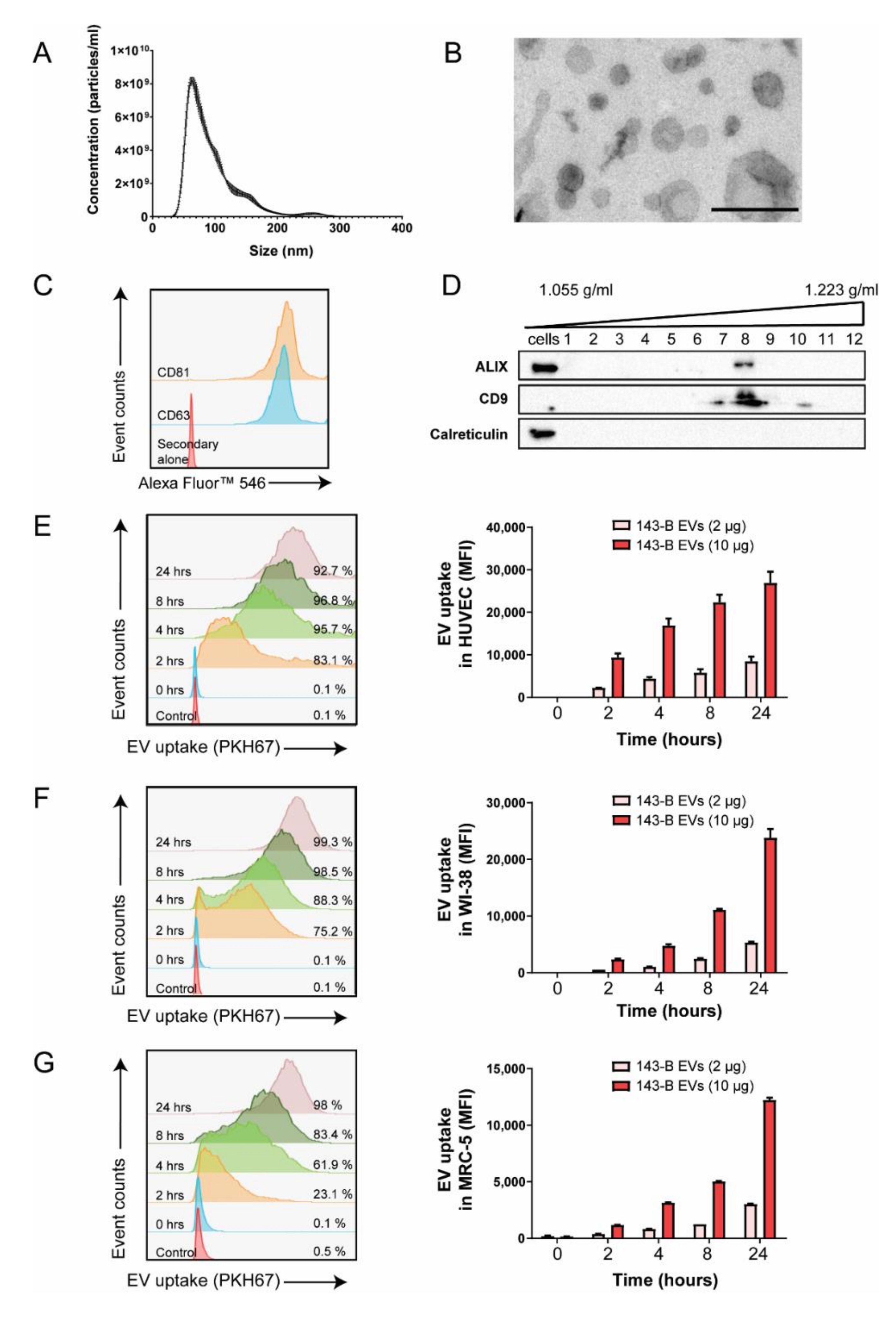
2.1. Osteosarcoma Cell-Derived Vesicles are Internalized by Endothelial Cells and Lung Fibroblasts In Vitro
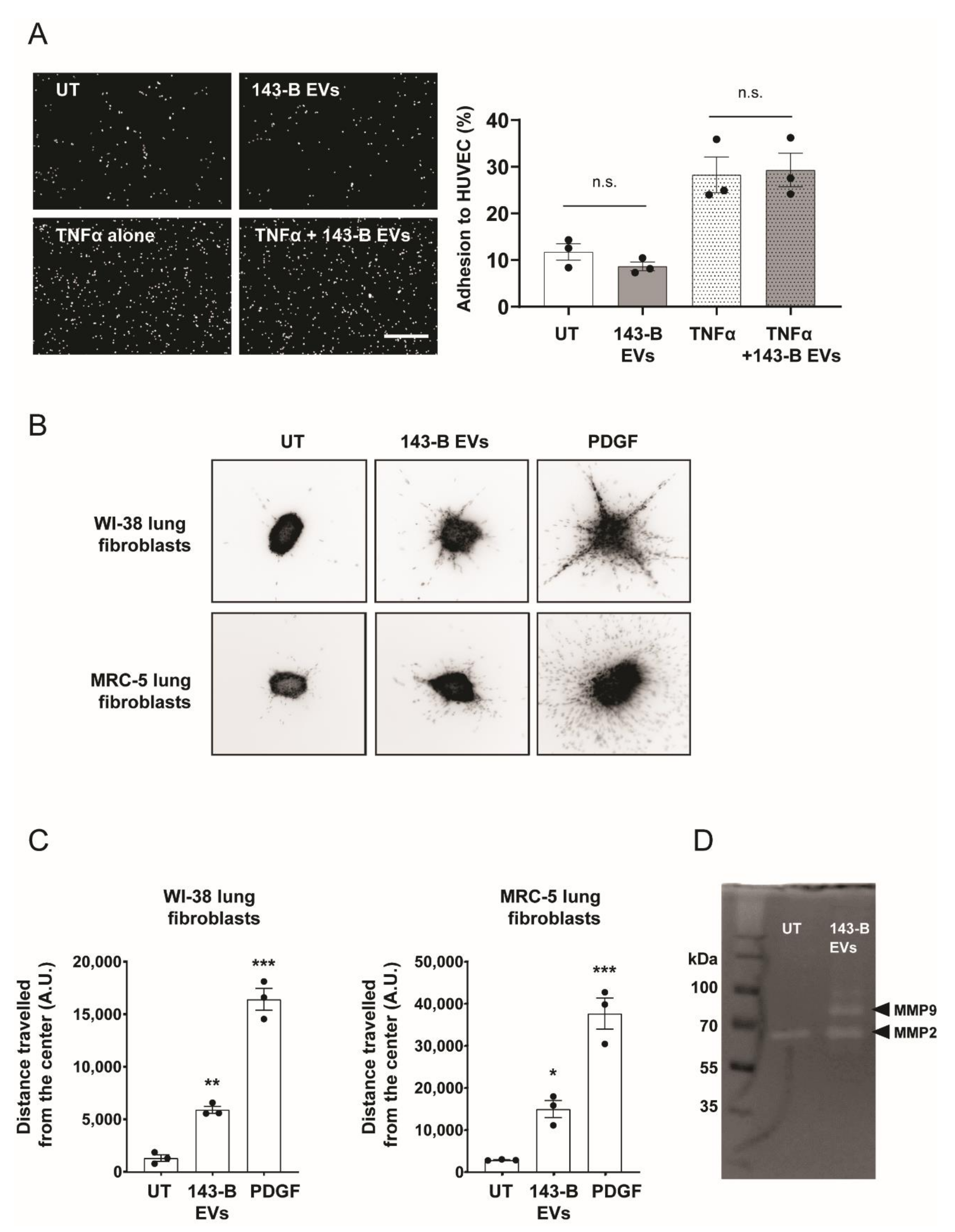
2.2. Osteosarcoma Cell-Derived Vesicles Promote the Functional Activation of Lung Fibroblasts but Not Endothelial Cells In Vitro
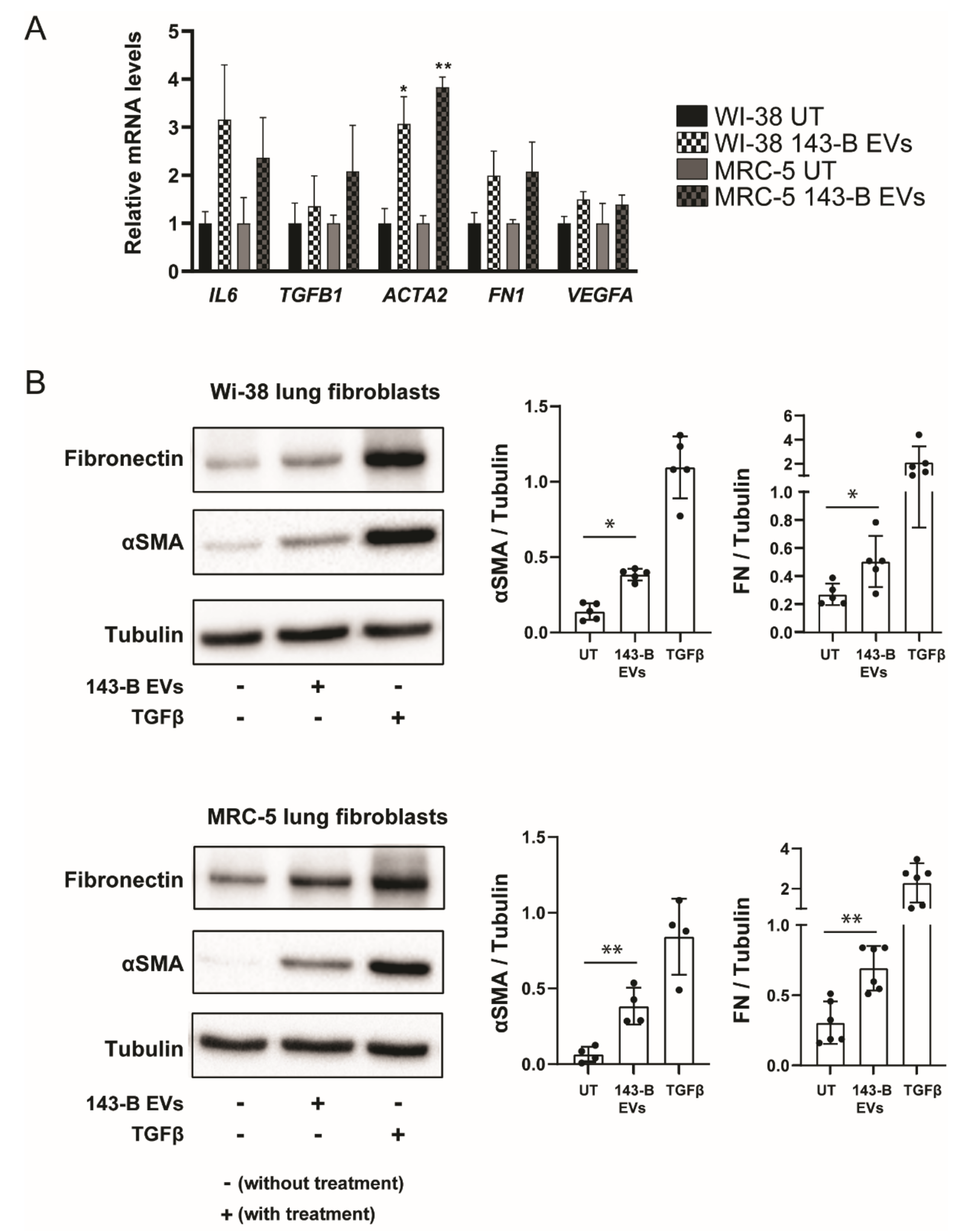
2.3. Osteosarcoma Cell-Derived Vesicles Cause Differentiation of Lung Fibroblasts into Myofibroblasts/Cancer-Associated Fibroblasts
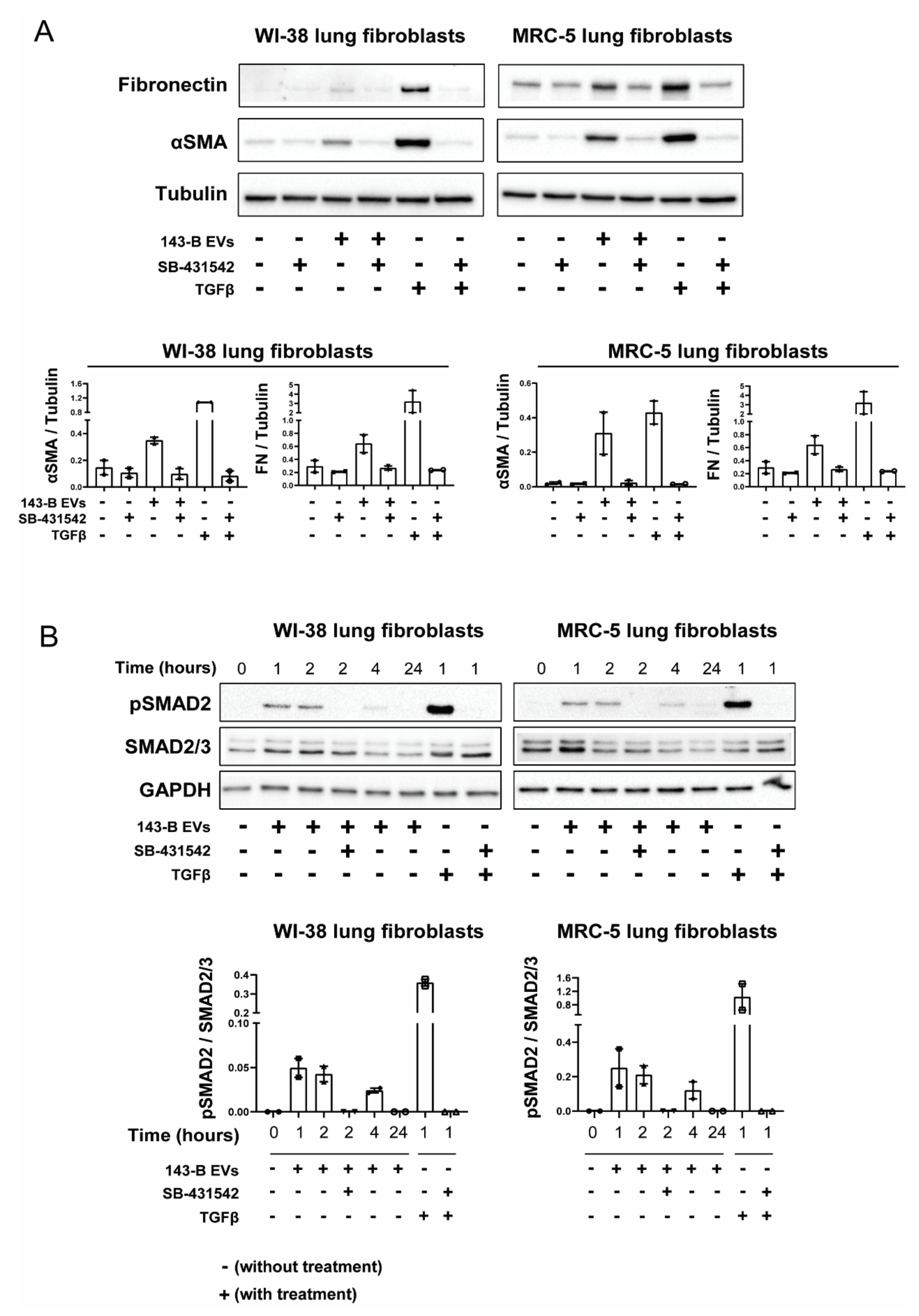
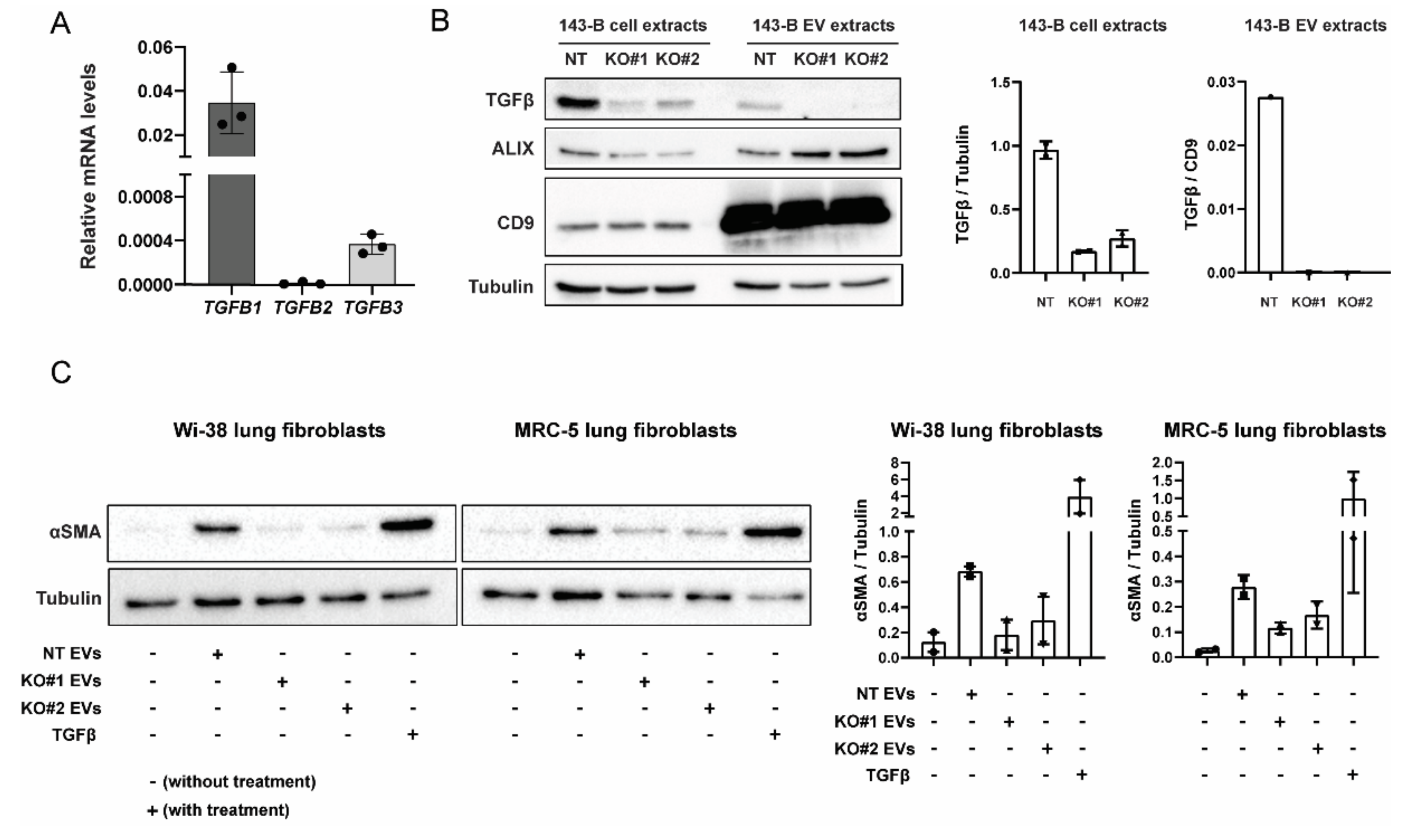
2.4. EV-Associated TGFβ Signaling Causes Myofibroblast Differentiation
2.5. CRISPR-Cas9-Mediated Deletion of TGFB1 Prevents Osteosarcoma-Derived EVs from Inducing Myofibroblast Differentiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. EV Isolation, Fibercell Hollow-Fiber Bioreactor Culture and Bioreactor EV Purification
4.3. Iodixanol Density Gradient Centrifugation
4.4. Nanoparticle Tracking Analysis (NTA)
4.5. Flow Cytometry Analysis of EV-Coated Beads
4.6. EV Labelling and Uptake
4.7. Western Blotting and Zymograhy Analysis
4.8. Negative Staining Transmission Electron Microscopy
4.9. RNA Isolation, Reverse Transcription and qRT-PCR
4.10. Adhesion Assay
4.11. Spheroid Invasion Assay
4.12. Generation of CRISPR/Cas9-Mediated Knockout Cells
4.13. ELISA
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EVs | Extracellular vesicles |
| CAFs | Cancer-associated fibroblasts |
| PMN | Pre-metastatic niche |
| NTA | Nanoparticle tracking analysis |
| SEM | Standard error of the mean |
| HUVEC | Human umbilical vein endothelial cells |
| ECM | Extracellular matrix |
| MFI | Mean fluorescence intensity |
| qRT-PCR | Real-time quantitative reverse transcription-polymerase chain reaction |
References
- Picci, P. Osteosarcoma (osteogenic sarcoma). Orphanet J. Rare Dis. 2007, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Chicon-Bosch, M.; Tirado, O.M. Exosomes in Bone Sarcomas: Key Players in Metastasis. Cells 2020, 9, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garimella, R.; Washington, L.; Isaacson, J.; Vallejo, J.; Spence, M.; Tawfik, O.; Rowe, P.; Brotto, M.; Perez, R. Extracellular Membrane Vesicles Derived from 143B Osteosarcoma Cells Contain Pro-Osteoclastogenic Cargo: A Novel Communication Mechanism in Osteosarcoma Bone Microenvironment. Transl. Oncol. 2014, 7, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raimondi, L.; De Luca, A.; Gallo, A.; Costa, V.; Russelli, G.; Cuscino, N.; Manno, M.; Raccosta, S.; Carina, V.; Bellavia, D.; et al. Osteosarcoma cell-derived exosomes affect tumor microenvironment by specific packaging of microRNAs. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Bao, Q.; Hu, C.; Wang, J.; Zhou, Q.; Wei, L.; Tong, L.; Zhang, W.; Shen, Y. Exosomal miR-675 from metastatic osteosarcoma promotes cell migration and invasion by targeting CALN1. Biochem. Biophys. Res. Commun. 2018, 500, 170–176. [Google Scholar] [CrossRef]
- Baglio, S.R.; Lagerweij, T.; Perez-Lanzon, M.; Ho, X.D.; Leveille, N.; Melo, S.A.; Cleton-Jansen, A.M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin. Cancer. Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef] [Green Version]
- Wolf-Dennen, K.; Gordon, N.; Kleinerman, E.S. Exosomal communication by metastatic osteosarcoma cells modulates alveolar macrophages to an M2 tumor-promoting phenotype and inhibits tumoricidal functions. OncoImmunology 2020, 9, 1747677. [Google Scholar] [CrossRef] [Green Version]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, L.T.; Peng, B.; Zhang, D.X.; Ma, V.; Mathey-Andrews, C.A.; Lam, C.K.; Kiomourtzis, T.; Jin, J.; McReynolds, L.; Huang, L. Tumor-secreted extracellular vesicles promote the activation of cancer-associated fibroblasts via the transfer of microRNA-125b. J. Extracell. Vesicles 2019, 8, 1599680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macklin, R.; Wang, H.; Loo, D.; Martin, S.; Cumming, A.; Cai, N.; Lane, R.; Ponce, N.S.; Topkas, E.; Inder, K. Extracellular vesicles secreted by highly metastatic clonal variants of osteosarcoma preferentially localize to the lungs and induce metastatic behaviour in poorly metastatic clones. Oncotarget 2016, 7, 43570. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.; Gui, J.; Zahedi, F.; Yu, P.; Cho, C.; Bhattacharya, S.; Carbone, C.J.; Yu, Q.; Katlinski, K.V.; Katlinskaya, Y.V.; et al. An Interferon-Driven Oxysterol-Based Defense against Tumor-Derived Extracellular Vesicles. Cancer Cell 2019, 35, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFbeta Signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z. Melanoma cell-secreted exosomal miR-155-5p induce proangiogenic switch of cancer-associated fibroblasts via SOCS1/JAK2/STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2018, 37, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Pasche, B.; Lee, C.; Grippo, P.J. TGF-β: Duality of function between tumor prevention and carcinogenesis. JNCI J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Lamora, A.; Talbot, J.; Mullard, M.; Brounais-Le Royer, B.; Redini, F.; Verrecchia, F. TGF-beta Signaling in Bone Remodeling and Osteosarcoma Progression. J. Clin. Med. 2016, 5, 96. [Google Scholar] [CrossRef]
- Franchi, A.; Arganini, L.; Baroni, G.; Calzolari, A.; Capanna, R.; Campanacci, D.; Caldora, P.; Masi, L.; Brandi, M.L.; Zampi, G. Expression of transforming growth factor β isoforms in osteosarcoma variants: Association of tgfβ1 with high-grade osteosarcomas. J. Pathol. J. Pathol. Soc. Great Br. Irel. 1998, 185, 284–289. [Google Scholar] [CrossRef]
- Lamora, A.; Talbot, J.; Bougras, G.; Amiaud, J.; Leduc, M.; Chesneau, J.; Taurelle, J.; Stresing, V.; Le Deley, M.C.; Heymann, M.F. Overexpression of smad7 blocks primary tumor growth and lung metastasis development in osteosarcoma. Clin. Cancer Res. 2014, 20, 5097–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamora, A.; Mullard, M.; Amiaud, J.; Brion, R.; Heymann, D.; Redini, F.; Verrecchia, F. Anticancer activity of halofuginone in a preclinical model of osteosarcoma: Inhibition of tumor growth and lung metastases. Oncotarget 2015, 6, 14413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Perut, F.; Roncuzzi, L.; Zini, N.; Massa, A.; Baldini, N. Extracellular nanovesicles secreted by human osteosarcoma cells promote angiogenesis. Cancers 2019, 11, 779. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsig, L.; Wang, L.; Cavalcante, M.C.; Cardilo-Reis, L.; Ferreira, P.L.; Mourão, P.A.; Esko, J.D.; Pavão, M.S. Selectin blocking activity of a fucosylated chondroitin sulfate glycosaminoglycan from sea cucumber effect on tumor metastasis and neutrophil recruitment. J. Biol. Chem. 2007, 282, 14984–14991. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Yamamoto, Y.; Sakamoto, N.; Shimomura, I.; Kogure, A.; Kumazaki, M.; Yokoi, A.; Yashiro, M.; Kiyono, T.; Yanagihara, K.; et al. Cancer extracellular vesicles contribute to stromal heterogeneity by inducing chemokines in cancer-associated fibroblasts. Oncogene 2019, 38, 5566–5579. [Google Scholar] [CrossRef]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 1–29. [Google Scholar] [CrossRef]
- Robl, B.; Botter, S.M.; Pellegrini, G.; Neklyudova, O.; Fuchs, B. Evaluation of intraarterial and intravenous cisplatin chemotherapy in the treatment of metastatic osteosarcoma using an orthotopic xenograft mouse model. J. Exp. Clin. Cancer Res. 2016, 35, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robl, B.; Botter, S.M.; Boro, A.; Meier, D.; Neri, D.; Fuchs, B. Evaluation of F8-TNF-alpha in Models of Early and Progressive Metastatic Osteosarcoma. Transl. Oncol. 2017, 10, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Gvozdenovic, A.; Arlt, M.J.; Campanile, C.; Brennecke, P.; Husmann, K.; Li, Y.; Born, W.; Muff, R.; Fuchs, B. CD44 enhances tumor formation and lung metastasis in experimental osteosarcoma and is an additional predictor for poor patient outcome. J. Bone Miner. Res. 2013, 28, 838–847. [Google Scholar] [CrossRef]
- Arlt, M.; Kopitz, C.; Pennington, C.; Watson, K.L.; Krell, H.-W.; Bode, W.; Gansbacher, B.; Khokha, R.; Edwards, D.R.; Krüger, A. Increase in gelatinase-specificity of matrix metalloproteinase inhibitors correlates with antimetastatic efficacy in a T-cell lymphoma model. Cancer Res. 2002, 62, 5543–5550. [Google Scholar]
- Brennecke, P.; Arlt, M.J.; Muff, R.; Campanile, C.; Gvozdenovic, A.; Husmann, K.; Holzwarth, N.; Cameroni, E.; Ehrensperger, F.; Thelen, M.; et al. Expression of the chemokine receptor CXCR7 in CXCR4-expressing human 143B osteosarcoma cells enhances lung metastasis of intratibial xenografts in SCID mice. PLoS ONE 2013, 8, e74045. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.S.; Pillong, M.; Kunze, J.; Burghardt, I.; Weller, M.; Grotzer, M.A.; Schneider, G.; Baumgartner, M. Computer-assisted quantification of motile and invasive capabilities of cancer cells. Sci. Rep. 2015, 5, 15338. [Google Scholar] [CrossRef] [Green Version]
- Naito, Y.; Hino, K.; Bono, H.; Ui-Tei, K. CRISPRdirect: Software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics 2015, 31, 1120–1123. [Google Scholar] [CrossRef]
- Morgens, D.W.; Wainberg, M.; Boyle, E.A.; Ursu, O.; Araya, C.L.; Tsui, C.K.; Haney, M.S.; Hess, G.T.; Han, K.; Jeng, E.E.; et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017, 8, 15178. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazumdar, A.; Urdinez, J.; Boro, A.; Migliavacca, J.; Arlt, M.J.E.; Muff, R.; Fuchs, B.; Snedeker, J.G.; Gvozdenovic, A. Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming. Int. J. Mol. Sci. 2020, 21, 5451. https://doi.org/10.3390/ijms21155451
Mazumdar A, Urdinez J, Boro A, Migliavacca J, Arlt MJE, Muff R, Fuchs B, Snedeker JG, Gvozdenovic A. Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming. International Journal of Molecular Sciences. 2020; 21(15):5451. https://doi.org/10.3390/ijms21155451
Chicago/Turabian StyleMazumdar, Alekhya, Joaquin Urdinez, Aleksandar Boro, Jessica Migliavacca, Matthias J.E. Arlt, Roman Muff, Bruno Fuchs, Jess Gerrit Snedeker, and Ana Gvozdenovic. 2020. "Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming" International Journal of Molecular Sciences 21, no. 15: 5451. https://doi.org/10.3390/ijms21155451
APA StyleMazumdar, A., Urdinez, J., Boro, A., Migliavacca, J., Arlt, M. J. E., Muff, R., Fuchs, B., Snedeker, J. G., & Gvozdenovic, A. (2020). Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming. International Journal of Molecular Sciences, 21(15), 5451. https://doi.org/10.3390/ijms21155451