Normoxic Tumour Extracellular Vesicles Modulate the Response of Hypoxic Cancer and Stromal Cells to Doxorubicin In Vitro

 ,
,  ,
,
Abstract
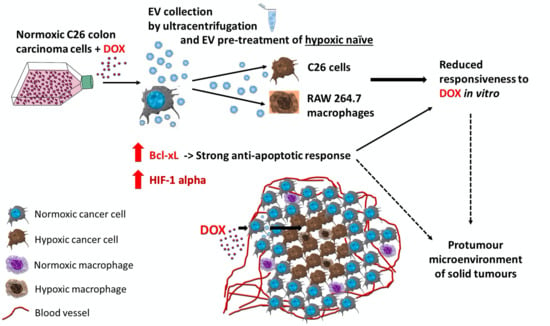
:
1. Introduction
2. Results
2.1. Under Normoxia, C26 Cells Were Less Responsive to DOX Compared to Hypoxia
2.2. Enhanced DOX Uptake in Hypoxic C26 Cells Compared to Its Uptake in Normoxic C26 Cells
2.3. Hypoxic Conditions Stimulated the Secretion of EVs by C26 Cells
2.4. TEV Effects on the Response of C26 Cells to DOX Administration
2.5. TEV Effects on the Response of the RAW 264.7 Macrophage-Like Cells to DOX Administration
2.6. Assessment of Resistance-Associated Markers in EV Donor C26 Cells
2.7. Assessment of Apoptotic Markers and HIF-1α in Normoxic TEV-Recipient C26 and RAW 264.7 Hypoxic Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Cell Viability Determination
4.3. Fluorescence Microscopy
4.4. Preparation of Pre-Spun Media for EV Harvesting
4.5. Isolation of EVs
4.6. Immunomagnetic Bead-Based Detection of EVs by FACS
4.7. Nanoparticle Tracking Analysis (NTA)
4.8. Assessment of the Effects of EVs Derived from DOX-Treated C26 Cells on the Response of C26 and RAW 264.7 Cells to DOX Treatment
4.9. Assessment of the Effects of C26 TEV and DOX-TEV Pretreatments on the Levels of Proteins Involved in Hypoxia Response and Apoptosis in Hypoxic C26 and RAW 264.7 Cells Subjected to DOX Treatment
4.10. Preparation of Cell Lysates
4.11. Western Blot Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EV(s) | extracellular vesicle(s) |
| TME | tumour microenvironment |
| TEV | tumour-derived extracellular vesicles |
| DOX | doxorubicin |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| Bcl-xL | B-cell lymphoma–extra-large anti-apoptotic protein |
| TAM | tumour-associated macrophages |
| HCR | hypoxia cytotoxicity ratio |
| IC50 | the half maximal inhibitory concentration |
| DIC | differential interference contrast |
| DAPI | 4′,6-diamidino-2-phenylindole |
| MFI | mean fluorescence intensity |
| NTA | nanoparticle tracking analysis |
| BAX | Bcl-2-associated X protein |
| AP-1 c-Jun | c-Jun subunit of activator protein 1 |
| c-Myc | c-master regulator of cell cycle entry and proliferative metabolism |
| NF-κB p65 | p65 subunit of nuclear factor kappa-light-chain-enhancer of activated B cells |
| PI3K | phosphatidylinositol-3 kinase |
| Akt | protein kinase B |
| c-Src | proto-oncogene tyrosine-protein kinase Src |
| RPMI | Roswell Park Memorial Institute |
| DMEM | Dulbecco’s modified Eagle’s medium |
| FBS | foetal bovine serum |
| MTS | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt) |
| PMS | phenazine methosulfate |
| PBS | phosphate-buffered saline |
| FACS | fluorescence-activated cell sorting |
| EDTA | ethylenediaminetetraacetic acid |
| RIPA | radioimmunoprecipitation assay buffer |
References
- Casazza, A.; Di Conza, G.; Wenes, M.; Finisguerra, V.; Deschoemaeker, S.; Mazzone, M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene 2013, 33, 1743–1754. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Lee, H.; Hong, I.-S. Double-edged sword of mesenchymal stem cells: Cancer-promoting versus therapeutic potential. Cancer Sci. 2017, 108, 1939–1946. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef]
- Murdoch, C.; Lewis, C.E. Macrophage migration and gene expression in response to tumor hypoxia. Int. J. Cancer 2005, 117, 701–708. [Google Scholar] [CrossRef]
- Van Overmeire, E.; Laoui, D.; Keirsse, J.; Van Ginderachter, J. Hypoxia and tumor-associated macrophages. OncoImmunology 2014, 3, e27561. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Lewis, C.E. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Patras, L.; Sesarman, A.; Licarete, E.; Luca, L.; Alupei, M.C.; Rakosy-Tican, E.; Banciu, M. Dual role of macrophages in the response of C26 colon carcinoma cells to 5-fluorouracil administration. Oncol. Lett. 2016, 12, 1183–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brücher, B.L.D.M.; Jamall, I.S. Cell-Cell Communication in the Tumor Microenvironment, Carcinogenesis, and Anticancer Treatment. Cell. Physiol. Biochem. 2014, 34, 213–243. [Google Scholar] [CrossRef] [PubMed]
- Dioufa, N.; Clark, A.M.; Ma, B.; Beckwitt, C.H.; Wells, A. Bi-directional exosome-driven intercommunication between the hepatic niche and cancer cells. Mol. Cancer 2017, 16, 172. [Google Scholar] [CrossRef] [Green Version]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Extracellular vesicles: Masters of intercellular communication and potential clinical interventions. J. Clin. Investig. 2016, 126, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Yuana, Y.; Sturk, A.; Nieuwland2R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.; Yang, F.; Miao, S.; Liu, W.; Wang, C.; Shu, Y.; Shen, H. Role of hypoxia-induced exosomes in tumor biology. Mol. Cancer 2018, 17, 120. [Google Scholar] [CrossRef]
- Kanada, M.; Bachmann, M.H.; Contag, C.H. Signaling by Extracellular Vesicles Advances Cancer Hallmarks. Trends Cancer 2016, 2, 84–94. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Tomasetti, M.; Lee, W.; Santarelli, L.; Neuzil, J. Exosome-derived microRNAs in cancer metabolism: Possible implications in cancer diagnostics and therapy. Exp. Mol. Med. 2017, 49, e285. [Google Scholar] [CrossRef] [Green Version]
- Carretero-González, A.; Otero, I.; Carril-Ajuria, L.; De Velasco, G.; Manso, L. Exosomes: Definition, Role in Tumor Development and Clinical Implications. Cancer Microenviron. 2018, 11, 13–21. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patras, L.; Banciu, M. Intercellular Crosstalk Via Extracellular Vesicles in Tumor Milieu as Emerging Therapies for Cancer Progression. Curr. Pharm. Des. 2019, 25, 1980–2006. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, A.M.; Fostel, J.M.; To, K.K.; Salcido, C.D.; Martin, S.E.; Chewning, K.J.; Wu, C.-P.; Varticovski, L.; Bates, S.E.; Caplen, N.J.; et al. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br. J. Cancer 2008, 98, 1515–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubertin, K.; Silva, A.K.; Luciani, N.; Espinosa, A.; Djemat, A.; Charue, D.; Gallet, F.; Blanc-Brude, O.; Wilhelm, C. Massive release of extracellular vesicles from cancer cells after photodynamic treatment or chemotherapy. Sci. Rep. 2016, 6, 35376. [Google Scholar] [CrossRef]
- Berghaus, L.J.; Moore, J.N.; Hurley, D.J.; Vandenplas, M.L.; Fortes, B.P.; Wolfert, M.A.; Boons, G.-J. Innate immune responses of primary murine macrophage-lineage cells and RAW 264.7 cells to ligands of Toll-like receptors 2, 3, and 4. Comp. Immunol. Microbiol. Infect. Dis. 2010, 33, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed M1 and M2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Y.; Zhang, F.; Zhao, Q.; Zhong, H. Increased anti-tumour activity by exosomes derived from doxorubicin-treated tumour cells via heat stress. Int. J. Hyperth. 2015, 31, 498–506. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nature 2018, 21, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Jabalee, J.; Towle, R.; Garnis, C. The Role of Extracellular Vesicles in Cancer: Cargo, Function, and Therapeutic Implications. Cells 2018, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour Cell Secretome in Chemoresistance and Tumour Recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.F.; Zhu, H.; Millard, R.W.; Fan, G.-C. Exosomes Function in Pro- and Anti-Angiogenesis. Curr. Angiogenesis 2013, 2, 54–59. [Google Scholar]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Tang, M.K.S.; Yue, P.Y.K.; Ip, P.P.C.; Huang, R.L.; Lai, H.-C.; Cheung, A.; Tse, K.Y.; Ngan, H.Y.; Wong, A.S.T. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat. Commun. 2018, 9, 2270. [Google Scholar] [CrossRef]
- Shao, Y.; Chen, T.; Zheng, X.; Yang, S.; Xu, K.; Chen, X.; Xu, F.; Wang, L.; Shen, Y.; Wang, T.; et al. Colorectal cancer-derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis 2018, 39, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Ciravolo, V.; Huber, V.; Ghedini, G.C.; Venturelli, E.; Morelli, D.; Villa, A.; Della Mina, P.; Filipazzi, P.; Pupa, S.M.; Bianchi, F.; et al. Potential role of HER2?overexpressing exosomes in countering trastuzumab?based therapy†. J. Cell. Physiol. 2011, 227, 658–667. [Google Scholar] [CrossRef]
- Giallombardo, M.; Taverna, S.; Alessandro, R.; Hong, D.; Rolfo, C. Exosome-mediated drug resistance in cancer: The near future is here. Ther. Adv. Med. Oncol. 2016, 8, 320–322. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Ying, X.; Wu, Q.; Wu, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wang, X. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget 2016, 7, 43076–43087. [Google Scholar] [CrossRef] [Green Version]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Müller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Möller, A. Breast Cancer-Derived Exosomes Alter Macrophage Polarization via gp130/STAT3 Signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2016, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, M.S.; Roy, A.; Rajpoot, S.; Liu, D.; Savai, R.; Banerjee, S.; Kawada, M.; Faisal, S.M.; Saluja, R.; Saqib, U.; et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflamm. Res. 2020, 69, 435–451. [Google Scholar] [CrossRef]
- Baj-Krzyworzeka, M.; Szatanek, R.; Węglarczyk, K.; Baran, J.; Urbanowicz, B.; Brański, P.; Ratajczak, M.Z.; Zembala, M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol. Immunother. 2005, 55, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Popēna, I.; Ābols, A.; Saulīte, L.; Pleiko, K.; Zandberga, E.; Jēkabsons, K.; Endzeliņš, E.; Llorente, A.; Linē, A.; Riekstina, U. Effect of colorectal cancer-derived extracellular vesicles on the immunophenotype and cytokine secretion profile of monocytes and macrophages. Cell Commun. Signal. 2018, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, D.; Lima, R.T.; Vasconcelos, M.H. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol. Med. 2015, 21, 595–608. [Google Scholar] [CrossRef]
- Sullivan, R.; Frederiksen, L.J.; Semenza, G.L.; Graham, C.H.; Paré, G.C. Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 2008, 7, 1961–1973. [Google Scholar] [CrossRef] [Green Version]
- Kibria, G.; Hatakeyama, H.; Akiyama, K.; Hida, K.; Harashima, H. Comparative Study of the Sensitivities of Cancer Cells to Doxorubicin, and Relationships between the Effect of the Drug-Efflux Pump P-gp. Boil. Pharm. Bull. 2014, 37, 1926–1935. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, M.; Ahmadihosseini, Z.; Allison, S.J.; Begum, S.; Rockley, K.; Sadiq, M.; Chintamaneni, S.; Lokwani, R.; Hughes, N.; Phillips, R.M. Hypoxia modulates the activity of a series of clinically approved tyrosine kinase inhibitors. Br. J. Pharmacol. 2013, 171, 224–236. [Google Scholar] [CrossRef]
- Pinzon-Daza, M.L.; Cuellar-Saenz, Y.; Nualart, F.; Ondo-Méndez, A.; Del Riesgo, L.; Castillo-Rivera, F.; Garzòn, R. Oxidative Stress Promotes Doxorubicin-Induced Pgp and BCRP Expression in Colon Cancer Cells Under Hypoxic Conditions. J. Cell. Biochem. 2017, 118, 1868–1878. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Berghe, T.V.; Krysko, D.V.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Sonowal, H.; Pal, P.B.; Wen, J.; Awasthi, S.; Ramana, K.V. Aldose reductase inhibitor increases doxorubicin-sensitivity of colon cancer cells and decreases cardiotoxicity. J. Diabetes Res. 2017, 7, 3183. [Google Scholar] [CrossRef]
- Endzelinš, E.; Ābols, A.; Bušs, A.; Zandberga, E.; Palviainen, M.; Siljander, P.; Linē, A. Extracellular vesicles derived from hypoxic colorectal cancer cells confer metastatic phenotype to non-metastatic cancer cells. Anticancer Res. 2018, 38, 5139–5147. [Google Scholar] [CrossRef]
- Lowry, M.C.; O’Driscoll, L. Can hi-jacking hypoxia inhibit extracellular vesicles in cancer? Drug Discov. Today 2018, 23, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Bandari, S.K.; Purushothaman, A.; Ramani, V.C.; Brinkley, G.J.; Chandrashekar, D.S.; Varambally, S.; Mobley, J.A.; Zhang, Y.; Brown, E.E.; Vlodavsky, I.; et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2018, 65, 104–118. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Topham, C.; Tighe, A.; Ly, P.; Bennett, A.; Sloss, O.; Nelson, L.; Ridgway, R.A.; Huels, D.; Littler, S.; Schandl, C.; et al. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell 2015, 28, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Trisciuoglio, D.; Tupone, M.G.; Desideri, M.; Di Martile, M.; Gabellini, C.; Buglioni, S.; Pallocca, M.; Alessandrini, G.; D’Aguanno, S.; Del Bufalo, D. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 2017, 8, 3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, U.H.; Landon, C.D.; Nien, C.Y.-C.; Hanna, G.; Rich, J.N.; Provenzale, J.M.; et al. Tumor cells upregulate normoxic HIF-1α in response to doxorubicin. Cancer Res. 2013, 73, 6230–6242. [Google Scholar] [CrossRef] [Green Version]
- Song, X.-R.; Liu, X.; Chi, W.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1α gene. Cancer Chemother. Pharmacol. 2006, 58, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Nardinocchi, L.; Puca, R.; Sacchi, A.; D’Orazi, G. Inhibition of HIF-1alpha activity by homeodomain-interacting protein kinase-2 correlates with sensitization of chemoresistant cells to undergo apoptosis. Mol. Cancer 2009, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Vardaki, I.; Sanchez, C.; Fonseca, P.; Olsson, M.; Chioureas, D.; Rassidakis, G.; Ullén, A.; Zhivotovsky, B.; Björkholm, M.; Panaretakis, T. Caspase-3–dependent cleavage of Bcl-xL in the stroma exosomes is required for their uptake by hematological malignant cells. Blood 2016, 128, 2655–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonafede, R.; Brandi, J.; Manfredi, M.; Scambi, I.; Schiaffino, L.; Merigo, F.; Turano, E.; Bonetti, B.; Marengo, E.; Cecconi, D.; et al. The Anti-Apoptotic Effect of ASC-Exosomes in an In Vitro ALS Model and Their Proteomic Analysis. Cells 2019, 8, 1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.; Hou, M.; Guan, Y.-S.; Jiang, M.; Yang, Y.; Gou, H. Expression of HIF-1alpha and VEGF in colorectal cancer: Association with clinical outcomes and prognostic implications. BMC Cancer 2009, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.E.; Imtiyaz, H.Z.; Sivanand, S.; Durham, A.C.; Skuli, N.; Hsu, S.; Mucaj, V.; Eisinger-Mathason, T.; Krock, B.L.; Giannoukos, D.N.; et al. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinog. 2014, 35, 1067–1077. [Google Scholar] [CrossRef]
- Lucchetti, D.; Ricciardi-Tenore, C.; Colella, F.; Sgambato, A. Extracellular Vesicles and Cancer: A Focus on Metabolism, Cytokines, and Immunity. Cancers 2020, 12, 171. [Google Scholar] [CrossRef] [Green Version]
- Strese, S.; Fryknäs, M.; Larsson, R.; Gullbo, J. Effects of hypoxia on human cancer cell line chemosensitivity. BMC Cancer 2013, 13, 331. [Google Scholar] [CrossRef] [Green Version]
- Clayton, A.; Court, J.; Navabi, H.; Adams, M.; Mason, M.D.; Hobot, J.A.; Newman, G.R.; Jasani, B. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J. Immunol. Methods 2001, 247, 163–174. [Google Scholar] [CrossRef]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.L.; Dobson, P.J.; et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomed-Nanotechnol. 2011, 7, 780–788. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | 12 h Drug Treatment | 24 h Drug Treatment | ||||
|---|---|---|---|---|---|---|
| Normoxia IC50 | Hypoxia IC50 | HCR (N:H) | Normoxia IC50 | Hypoxia IC50 | HCR (N:H) | |
| DOX | 3.704 | 1.922 | 1.93 | 0.929 | 0.407 | 2.28 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patras, L.; Fens, M.H.A.M.; Vader, P.; Barendrecht, A.; Sesarman, A.; Banciu, M.; Schiffelers, R. Normoxic Tumour Extracellular Vesicles Modulate the Response of Hypoxic Cancer and Stromal Cells to Doxorubicin In Vitro. Int. J. Mol. Sci. 2020, 21, 5951. https://doi.org/10.3390/ijms21175951
Patras L, Fens MHAM, Vader P, Barendrecht A, Sesarman A, Banciu M, Schiffelers R. Normoxic Tumour Extracellular Vesicles Modulate the Response of Hypoxic Cancer and Stromal Cells to Doxorubicin In Vitro. International Journal of Molecular Sciences. 2020; 21(17):5951. https://doi.org/10.3390/ijms21175951
Chicago/Turabian StylePatras, Laura, Marcel H. A. M. Fens, Pieter Vader, Arjan Barendrecht, Alina Sesarman, Manuela Banciu, and Raymond Schiffelers. 2020. "Normoxic Tumour Extracellular Vesicles Modulate the Response of Hypoxic Cancer and Stromal Cells to Doxorubicin In Vitro" International Journal of Molecular Sciences 21, no. 17: 5951. https://doi.org/10.3390/ijms21175951
APA StylePatras, L., Fens, M. H. A. M., Vader, P., Barendrecht, A., Sesarman, A., Banciu, M., & Schiffelers, R. (2020). Normoxic Tumour Extracellular Vesicles Modulate the Response of Hypoxic Cancer and Stromal Cells to Doxorubicin In Vitro. International Journal of Molecular Sciences, 21(17), 5951. https://doi.org/10.3390/ijms21175951