Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities

 ,
,  ,
, 
 ,
,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Gastric Cancer
2.1. Anatomical, Histological, and Molecular Classification of Gastric Cancer
2.2. Gastric Cancer Clinical Management
3. Epigenetics of GC
3.1. DNA Methylation
3.2. Histone Modifications
3.2.1. Histone Methylation
3.2.2. Histone Acetylation
4. Current and New Epigenetic Strategies for Gastric Cancer Treatment
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Cancer Facts & Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Personal habits and indoor combustions. Volume 100 E. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 1–538. [Google Scholar]
- Plummer, M.; Franceschi, S.; Vignat, J.; Forman, D.; de Martel, C. Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 2015, 136, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, V.; Soleimanian, A.; Ebrahimi, T.; Azargun, R.; Yazdani, P.; Eyvazi, S.; Tarhriz, V. Epigenetic modifications in gastric cancer: Focus on DNA methylation. Gene 2020, 742, 144577. [Google Scholar] [CrossRef] [PubMed]
- Abdelfatah, E.; Kerner, Z.; Nanda, N.; Ahuja, N. Epigenetic therapy in gastrointestinal cancer: The right combination. Ther. Adv. Gastroenterol. 2016, 9, 560–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornschein, J.; Malfertheiner, P. Gastric carcinogenesis. Langenbeck’s Arch. Surg. 2011, 396, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar] [PubMed]
- Hayakawa, Y.; Fox, J.G.; Wang, T.C. The Origins of Gastric Cancer From Gastric Stem Cells. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 331–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.A.; McDonald, S.A.; Wright, N.A. Field cancerization in the GI tract. Future Oncol. 2011, 7, 981–993. [Google Scholar] [CrossRef]
- Baba, Y.; Ishimoto, T.; Kurashige, J.; Iwatsuki, M.; Sakamoto, Y.; Yoshida, N.; Watanabe, M.; Baba, H. Epigenetic field cancerization in gastrointestinal cancers. Cancer Lett. 2016, 375, 360–366. [Google Scholar] [CrossRef]
- Sitaraman, R. Helicobacter pylori DNA methyltransferases and the epigenetic field effect in cancerization. Front. Microbiol. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Siewert, J.R.; Stein, H.J. Classification of adenocarcinoma of the oesophagogastric junction. Br. J. Surg. 1998, 85, 1457–1459. [Google Scholar] [PubMed]
- American Joint Committee on Cancer. JCC Cancer Staging Manual; Springer: New York, NY, USA, 2017. [Google Scholar]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Lauwers, G.; Carneiro, F.; Graham, D.Y. WHO classification of tumours of the digestive system-3rd chapter. In WHO Classifcation Tumours Dig Syst, 4th ed.; IARC: Lyon, France, 2010; pp. 44–58. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.-M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Lutz, M.; Zalcberg, J.R.; Ducreux, M.; Ajani, J.A.; Allum, W.; Aust, D.; Bang, Y.-J.; Cascinu, S.; Hölscher, A.; Jankowski, J.; et al. Highlights of the EORTC St. Gallen International Expert Consensus on the primary therapy of gastric, gastroesophageal and oesophageal cancer-differential treatment strategies for subtypes of early gastroesophageal cancer. Eur. J. Cancer 2012, 48, 2941–2953. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Dicato, M.; Geva, R.; Arber, N.; Bang, Y.; Benson, A.; Cervantes, A.; Diaz-Rubio, E.; Ducreux, M.; Glynne-Jones, R.; et al. The diagnosis and management of gastric cancer: Expert discussion and recommendations from the 12th ESMO/World Congress on Gastrointestinal Cancer, Barcelona, 2010. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22 (Suppl. 5), v1–v9. [Google Scholar] [CrossRef]
- Schmidt, B.; Yoon, S.S. D1 versus D2 lymphadenectomy for gastric cancer. J. Surg. Oncol. 2013, 107, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.D.; Unverzagt, S.; Grothe, W.; Kleber, G.; Grothey, A.; Haerting, J.; Fleig, W.E. Chemotherapy for advanced gastric cancer. Cochrane Database Syst. Rev. 2010, CD004064. [Google Scholar] [CrossRef]
- Glimelius, B.; Ekström, K.; Hoffman, K.; Graf, W.; Sjödén, P.-O.; Haglund, U.; Svensson, C.; Enander, L.-K.; Linné, T.; Sellsröm, H.; et al. Randomized comparison between chemotherapy plus best supportive care with best supportive care in advanced gastric cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1997, 8, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Moiseyenko, V.M.; Tjulandin, S.; Majlis, A.; Constenla, M.; Boni, C.; Rodrigues, A.; Fodor, M.; Chao, Y.; Voznyi, E.; et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: A report of the V325 Study Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 4991–4997. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.-J.; Iwasa, S.; Sugimoto, N.; Ryu, M.-H.; Sakai, D.; Chung, H.-C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N. Engl. J. Med. 2020, 382, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Santos, L.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef]
- Wilke, H.; Muro, K.; Van Cutsem, E.; Oh, S.-C.; Bodoky, G.; Shimada, Y.; Hironaka, S.; Sugimoto, N.; Lipatov, O.; Kim, T.-Y.; et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): A double-blind, randomised phase 3 trial. Lancet Oncol. 2014, 15, 1224–1235. [Google Scholar] [CrossRef]
- Lazar, D.C.; Taban, S.; Cornianu, M.; Faur, A.; Goldis, A. New advances in targeted gastric cancer treatment. World J. Gastroenterol. 2016, 22, 6776–6799. [Google Scholar] [CrossRef]
- Song, H.; Zhu, J.; Lu, D.; Song, H.; Zhu, J.; Lu, D. Molecular-targeted first-line therapy for advanced gastric cancer. Cochrane Database Syst. Rev. 2016, 7, CD011461. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Y.; Wang, H. Use of immunotherapy in the treatment of gastric cancer (Review). Oncol. Lett. 2019, 18, 5681–5690. [Google Scholar] [CrossRef]
- Coutzac, C.; Pernot, S.; Chaput, N.; Zaanan, A. Critical Reviews in Oncology/Hematology Immunotherapy in advanced gastric cancer, is it the future ? Crit. Rev. Oncol./Hematol. 2019, 133, 25–32. [Google Scholar] [CrossRef]
- Li, Y.; Liang, J.; Hou, P. Hypermethylation in gastric cancer. Clin. Chim. Act. 2015, 448, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Yoda, Y.; Takeshima, H.; Niwa, T.; Kim, J.G.; Ando, T.; Kushima, R.; Sugiyama, T.; Katai, H.; Noshiro, H.; Ushijima, T. Integrated analysis of cancer-related pathways affected by genetic and epigenetic alterations in gastric cancer. Gastric Cancer 2015, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wu, Q.; Wang, L.; Wang, H.; Yin, F. A DNA methylation signature to improve survival prediction of gastric cancer. Clin. Epigenet. 2020, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zheng, Y.; Pu, K.; Zhao, D.; Wang, Y.; Guan, Q.; Zhou, Y. A four-DNA methylation signature as a novel prognostic biomarker for survival of patients with gastric cancer. Cancer Cell Int. 2020, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wei, C.; Zhong, Y.; Zhang, Y.; Long, J.; Huang, S.; Xie, F.; Tian, Y.; Wang, X.; Zhao, H.; et al. Development and validation of a prognostic nomogram for gastric cancer based on DNA methylation-driven differentially expressed genes. Int. J. Biol. Sci. 2020, 16, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.P.; Kopetz, S.; Ajani, J.A.; Calin, G.A. Non-coding RNAs in GI cancers: From cancer hallmarks to clinical utility. Gut 2020, 69, 748–763. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Padmanabhan, N.; Ushijima, T.; Tan, P. How to stomach an epigenetic insult: The gastric cancer epigenome. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 467–478. [Google Scholar] [CrossRef]
- Matsusaka, K.; Kaneda, A.; Nagae, G.; Ushiku, T.; Kikuchi, Y.; Hino, R.; Uozaki, H.; Seto, Y.; Takada, K.; Aburatani, H.; et al. Classification of Epstein-Barr virus-positive gastric cancers by definition of DNA methylation epigenotypes. Cancer Res. 2011, 71, 7187–7197. [Google Scholar] [CrossRef] [Green Version]
- Corso, G.; Carvalho, J.; Marrelli, D.; Vindigni, C.; Carvalho, B.; Seruca, R.; Roviello, F.; Oliveira, C. Somatic mutations and deletions of the e-cadherin gene predict poor survival of patients with gastric cancer. J. Clin. Oncol. 2013, 31, 868–875. [Google Scholar] [CrossRef] [Green Version]
- van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.C.; Hardwick, R.H.; Ausems, M.G.E.M.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, J.C.; Oliveira, C.; Carvalho, R.; Soares, P.; Berx, G.; Caldas, C.; Seruca, R.; Carneiro, F.; Sobrinho-Simöes, M. E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene 2001, 20, 1525–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro, P.; Fernandes, M.S.; Figueiredo, J.; Caldeira, J.; Carvalho, J.; Pinheiro, H.; Leite, M.; Melo, S.; Oliveira, P.; Simões-Correia, J.; et al. E-cadherin dysfunction in gastric cancer—Cellular consequences, clinical applications and open questions. FEBS Lett. 2012, 586, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Shibata, T.; Arisawa, T.; Nakamura, M.; Yamashita, H.; Yoshioka, D.; Okubo, M.; Yonemura, J.; Maeda, Y.; Maruyama, N.; et al. CpG island promoter methylation (CIHM) status of tumor suppressor genes correlates with morphological appearances of gastric cancer. Anticancer Res. 2010, 30, 239–244. [Google Scholar] [PubMed]
- Balassiano, K.; Lima, S.; Jenab, M.; Overvad, K.; Tjonneland, A.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Canzian, F.; Kaaks, R.; Boeing, H.; et al. Aberrant DNA methylation of cancer-associated genes in gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Cancer Lett. 2011, 311, 85–95. [Google Scholar] [CrossRef]
- Shenoy, S. CDH1 (E-cadherin) mutation and gastric cancer: Genetics, molecular mechanisms and guidelines for management. Cancer Manag. Res. 2019, 11, 10477–10486. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chu, K.M. E-cadherin and gastric cancer: Cause, consequence, and applications. Biomed. Res. Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.O.O.; Wong, B.C.Y.; Lan, H.Y.; Loke, S.L.; Chan, W.K.; Hui, W.M.; Yuen, Y.H.; Ng, I.; Hou, L.; Wong, W.M.; et al. Deregulation of E-cadherin-catenin complex in precancerous lesions of gastric adenocarcinoma. J. Gastroenterol. Hepatol. 2003, 18, 534–539. [Google Scholar] [CrossRef]
- Chan, A.O.O.; Peng, J.Z.; Lam, S.K.; Lai, K.C.; Yuen, M.F.; Cheung, H.K.L.; Kwong, Y.L.; Rashid, A.; Chan, C.K.; Wong, B.C.-Y. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut 2006, 55, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Zhu, J.; Shan, L.; Han, Z.; Aerxiding, P.; Quhai, A.; Zeng, F.; Wang, Z.; Li, H. The clinicopathological significance of CDH1 in gastric cancer: A meta-analysis and systematic review. Drug Des. Devel. Ther. 2015, 9, 2149–2157. [Google Scholar] [CrossRef] [Green Version]
- Graziano, F.; Arduini, F.; Ruzzo, A.; Mandolesi, A.; Bearzi, I.; Silva, R.; Muretto, P.; Testa, E.; Mari, D.; Magnani, M.; et al. Combined analysis of E-cadherin gene (CDH1) promoter hypermethylation and E-cadherin protein expression in patients with gastric cancer: Implications for treatment with demethylating drugs. Ann Oncol. 2004, 15, 489–492. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Harfe, B.D.; Jinks-Robertson, S. Mismatch repair proteins and mitotic genome stability. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2000, 451, 151–167. [Google Scholar] [CrossRef]
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Lu, Y.; Herman, J.G.; Brock, M.V.; Zhao, P.; Guo, M. Predictive value of CHFR and MLH1 methylation in human gastric cancer. Gastric Cancer 2015, 18, 280–287. [Google Scholar] [CrossRef]
- Kitajima, Y.; Miyazaki, K.; Matsukura, S.; Tanaka, M.; Sekiguchi, M. Loss of expression of DNA repair enzymes MGMT, hMLH1, and hMSH2 during tumor progression in gastric cancer. Gastric Cancer 2003, 6, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Shigeyasu, K.; Nagasaka, T.; Mori, Y.; Yokomichi, N.; Kawai, T.; Fuji, T.; Kimura, K.; Umeda, Y.; Kagawa, S.; Goel, A.; et al. Clinical significance of MLH1 methylation and CpG island methylator phenotype as prognostic markers in patients with gastric cancer. PLoS ONE 2015, 10, e0130409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balgkouranidou, I.; Matthaios, D.; Karayiannakis, A.; Bolanaki, H.; Michailidis, P.; Xenidis, N.; Amarantidis, K.; Chelis, L.; Trypsianis, G.; Chatzaki, E.; et al. Prognostic role of APC and RASSF1A promoter methylation status in cell free circulating DNA of operable gastric cancer patients. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 778, 46–51. [Google Scholar] [CrossRef]
- Fang, J.Y.; Zhu, S.S.; Xiao, S.D.; Jiang, S.J.; Shi, Y.; Chen, X.Y.; Zhou, X.M.; Qian, L.F. Studies on the hypomethylation of c-myc, c-Ha-ras oncogenes and histopathological changes in human gastric carcinoma. J. Gastroenterol. Hepatol. 1996, 11, 1079–1082. [Google Scholar] [CrossRef]
- Yu, J.; Tao, Q.; Cheng, Y.Y.; Lee, K.Y.; Ng, S.S.M.; Cheung, K.F.; Tian, L.; Rha, S.Y.; Neumann, U.; Röcken, C.; et al. Promoter methylation of the Wnt/β-catenin signaling antagonist Dkk-3 is associated with poor survival in gastric cancer. Cancer 2009, 115, 49–60. [Google Scholar] [CrossRef]
- Ksiaa, F.; Ziadi, S.; Amara, K.; Korbi, S.; Trimeche, M. Biological significance of promoter hypermethylation of tumor-related genes in patients with gastric carcinoma. Clin. Chim. Acta 2009, 404, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.-B.; Qiu, X.-P.; Zhang, S.; Wang, C.; Zheng, F. CDKN2A promoter methylation and hepatocellular carcinoma risk: A meta-analysis. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Cai, W.; Shi, H.; Wang, Y.; Li, M.; Jiao, J.; Chen, M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: A meta-analysis. Br. J. Cancer 2013, 108, 2542–2548. [Google Scholar] [CrossRef]
- He, D.; Zhang, Y.-W.; Zhang, N.-N.; Zhou, L.; Chen, J.-N.; Jiang, Y.; Shao, C.-K. Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein–Barr virus-associated gastric carcinomas. Med. Oncol. 2015, 32, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.X.; Deng, D.J.; Pan, K.F.; Zhang, L.; Zhang, Y.; Zhou, J.; You, W.C. Promoter methylation of p16 associated with helicobacter pylori infection in precancerous gastric lesions: A population-based study. Int. J. Cancer 2009, 124, 434–439. [Google Scholar] [CrossRef]
- Ryan, J.L.; Jones, R.J.; Kenney, S.C.; Rivenbark, A.G.; Tang, W.; Knight, E.R.; Coleman, W.B.; Gulley, M.L. Epstein-Barr virus-specific methylation of human genes in gastric cancer cells. Infect. Agents Cancer 2010, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.M.; Kim, N.; Park, J.H.; Kang, G.H.; Kim, J.S.; Jung, H.C.; Song, I.S. Prediction of the risk for gastric cancer using candidate methylation markers in the non-neoplastic gastric mucosae. J. Pathol. 2012, 226, 654–665. [Google Scholar] [CrossRef]
- Abbaszadegan, M.R.; Moaven, O.; Sima, H.R.; Ghafarzadegan, K.; A’rabi, A.; Forghani, M.N.; Raziee, H.R.; Mashhadinejad, A.; Jafarzadeh, M.; Esmaili-Shandiz, E.; et al. p16 promoter hypermethylation: A useful serum marker for early detection of gastric cancer. World J. Gastroenterol. 2008, 14, 2055–2060. [Google Scholar] [CrossRef]
- Ichikawa, D.; Koike, H.; Ikoma, H.; Ikoma, D.; Tani, N.; Otsuji, E.; Kitamura, K.; Yamagishi, H. Detection of aberrant methylation as a tumor marker in serum of patients with gastric cancer. Anticancer Res. 2004, 24, 2477–2481. [Google Scholar] [PubMed]
- Pimson, C.; Ekalaksananan, T.; Pientong, C.; Promthet, S.; Putthanachote, N.; Suwanrungruang, K.; Wiangnon, S. Aberrant methylation of PCDH10 and RASSF1A genes in blood samples for non-invasive diagnosis and prognostic assessment of gastric cancer. PeerJ 2016, 4, e2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Ito, K.; Sakakura, C.; Fukamachi, H.; Inoue, K.-I.; Chi, X.Z.; Lee, K.Y.; Nomura, S.; Lee, C.W.; Han, S.B.; et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002, 109, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.Y.; Lee, H.J.; Hwang, K.S.; Lee, M.; Kim, J.W.; Bang, Y.J.; Kang, G.H. Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma. Lab. Investig. 2004, 84, 479–484. [Google Scholar] [CrossRef]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in Nucleic Acid Methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Howell, C.Y.; Bestor, T.H.; Ding, F.; Latham, K.E.; Mertineit, C.; Trasler, J.M.; Chaillet, J.R. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 2001, 104, 829–838. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Fattahi, S.; Kosari-Monfared, M.; Ghadami, E.; Golpour, M.; Khodadadi, P.; Ghasemiyan, M.; Niaki, H.-A. Infection-associated epigenetic alterations in gastric cancer: New insight in cancer therapy. J. Cell Physiol. 2018, 233, 9261–9270. [Google Scholar] [CrossRef]
- Yang, J.; Wei, X.; Wu, Q.; Xu, Z.; Gu, D.; Jin, Y.; Shen, Y.; Huang, H.; Fan, H.; Chen, J. Clinical significance of the expression of DNA methyltransferase proteins in gastric cancer. Mol. Med. Rep. 2011, 4, 1139–1143. [Google Scholar]
- Mutze, K.; Langer, R.; Schumacher, F.; Becker, K.; Ott, K.; Novotny, A.; Hapfelmeier, A.; Höfler, H.; Keller, G. DNA methyltransferase 1 as a predictive biomarker and potential therapeutic target for chemotherapy in gastric cancer. Eur. J. Cancer 2011, 47, 1817–1825. [Google Scholar] [CrossRef]
- Ma, T.; Li, H.; Sun, M.; Yuan, Y.; Sun, L.P. DNMT1 overexpression predicting gastric carcinogenesis, subsequent progression and prognosis: A meta and bioinformatic analysis. Oncotarget 2017, 8, 96396–96408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Jia, Z.; Cao, D.; Jin, M.S.; Kong, F.; Suo, J.; Cao, X. Polymorphisms of the DNA Methyltransferase 1 Associated with Reduced Risks of Helicobacter pylori Infection and Increased Risks of Gastric Atrophy. PLoS ONE 2012, 7, e46058. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Wu, X.; Cao, D.; Wang, C.; You, L.; Jin, M.; Wen, S.; Cao, X.; Jiang, J. Polymorphisms of the DNA Methyltransferase 1 Gene Predict Survival of Gastric Cancer Patients Receiving Tumorectomy. Dis. Mark. 2016, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Fan, H.; Liu, D.; Zhang, S.; Zhang, F.; Xu, H. DNMT3B promoter polymorphism and risk of gastric cancer. Dig. Dis. Sci. 2010, 55, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jia, Z.; Ma, H.; Cao, D.; Wu, X.; Wen, S.; You, L.; Cao, X.; Jiang, J. DNA methyltransferase 3a rs1550117 genetic polymorphism predicts poor survival in gastric cancer patients. Int. J. Clin. Exp. Pathol. 2015, 8, 14864–14874. [Google Scholar]
- Fan, H.; Liu, D.; Qiu, X.; Qiao, F.; Wu, Q.; Su, X.; Zhang, F.; Song, Y.; Zhao, Z. A functional polymorphism in the DNA methyltransferase-3A promoter modifies the susceptibility in gastric cancer but not in esophageal carcinoma. BMC Med. 2010, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, Z.; Zhang, L.; Gu, Y.; Ma, Y.; Li, X. Association of five genetic variations in DNMT1 and DNMT3A with gastric cancer in a Chinese population. Future Oncol. 2018, 14, 1731–1739. [Google Scholar] [CrossRef]
- Li, H.; Li, W.; Liu, S.; Zong, S.; Wang, W.; Ren, J.; Li, Q.; Hou, F.; Shi, Q. DNMT1, DNMT3A and DNMT3B Polymorphisms Associated With Gastric Cancer Risk: A Systematic Review and Meta-analysis. EBioMedicine 2016, 13, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Neves, M.; Ribeiro, J.; Medeiros, R.; Sousa, H. Genetic polymorphism in DNMTs and gastric cancer: A systematic review and meta-analysis. Porto Biomed. J. 2016, 1, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.M.; Szyf, M. Human DNA methyltransferase gene DNMT1 is regulated by the APC pathway evident in a variety of cancers, including colorectal cancer genes have been proposed, a causal downstream agent established. Because previous work implicates DNA methyl-epigenetic. Carcinogenesis 2003, 24, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, J.C.; Khan, S.A.; Christison-Lagay, E.R.; Cha, C. APC mutational patterns in gastric adenocarcinoma are enriched for missense variants with associated decreased survival. Genes Chromosom. Cancer 2020, 59, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiao, D.; Dou, M.; Zhang, W.; Hua, H.; Chen, J.; Li, Z.; Li, L.; Han, X. Association of APC gene promoter methylation and the risk of gastric cancer. Medicine 2020, 99, e19828. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, A.R. Helicobacter, Inflammation, and Gastric Cancer. Curr. Pathobiol. Rep. 2013, 1, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigier-Bompadre, M.; Moos, V.; Belogolova, E.; Allers, K.; Schneider, T.; Churin, Y.; Ignatius, R.; Meyer, T.F.; Aebischer, T. Modulation of the CD4 + T-cell response by helicobacter pylori depends on known virulence factors and bacterial cholesterol and cholesterol α-glucoside content. J. Infect. Dis. 2011, 204, 1339–1348. [Google Scholar] [CrossRef]
- Chang, C.C.; Kuo, W.S.; Chen, Y.C.; Perng, C.L.; Lin, H.J.; Ou, Y.H. Fragmentation of CagA reduces hummingbird phenotype induction by helicobactor pylori. PLoS ONE 2016, 11, e0150061. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Machado, J.C.; Leite, M.; Carneiro, F.; Figueiredo, C. The number of Helicobacter pylori CagA EPIYA C tyrosine phosphorylation motifs influences the pattern of gastritis and the development of gastric carcinoma. Histopathology 2012, 60, 992–998. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Yu, J.; Chan, M.T.V.; To, K.F.; Cheng, A.S.L. Combinatorial epigenetic deregulation by Helicobacter pylori and Epstein–Barr virus infections in gastric tumourigenesis. J. Pathol. 2016, 239, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, D.Q.; Gigek, C.O.; Chen, E.S.; Burbano, R.R.; Smith, M.D.A.C. DNA and histone methylation in gastric carcinogenesis. World J. Gastroenterol. 2013, 19, 1182–1192. [Google Scholar] [CrossRef]
- Fu, D.G. Epigenetic alterations in gastric cancer (Review). Mol. Med. Rep. 2015, 12, 3223–3230. [Google Scholar] [CrossRef] [Green Version]
- Rando, O.J. Combinatorial complexity in chromatin structure and function: Revisiting the histone code. Curr. Opin. Genet. Dev. 2012, 22, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: Targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Ji, G.; Gao, Z.; Han, X.; Ye, M.; Yuan, Z.; Luo, H.; Huang, X.; Natarajan, K.; Wang, J.; et al. Direct ChIP-bisulfite sequencing reveals a role of H3K27me3 mediating aberrant hypermethylation of promoter CpG islands in cancer cells. Genomics 2014, 103, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar]
- Park, Y.S.; Jin, M.Y.; Kim, Y.J.; Yook, J.H.; Kim, B.S.; Jang, S.J. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann. Surg. Oncol. 2008, 15, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.H.; Park, J.L.; Kim, M.; Kim, J.H.; Lee, H.C.; Kim, H.J.; Noh, S.M.; Song, K.S.; Yoo, H.S.; Paik, S.G.; et al. Aberrant up-regulation of LAMB3 and LAMC2 by promoter demethylation in gastric cancer. Biochem. Biophys. Res. Commun. 2011, 406, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, F.; Qi, G.; Yuan, S.; Zhang, G.; Tang, B.; He, S. KDM5B is overexpressed in gastric cancer and is required for gastric cancer cell proliferation and metastasis. Am. J. Cancer Res. 2015, 5, 87–100. [Google Scholar]
- Yang, H.; Liu, Z.; Yuan, C.; Zhao, Y.; Wang, L.; Hu, J.; Xie, D.; Wang, L.; Chen, D. Elevated JMJD1A is a novel predictor for prognosis and a potential therapeutic target for gastric cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11092–11099. [Google Scholar]
- Hu, C.E.; Liu, Y.C.; Zhang, H.D.; Huang, G.J. JMJD2A predicts prognosis and regulates cell growth in human gastric cancer. Biochem. Biophys. Res. Commun. 2014, 449, 1–7. [Google Scholar] [CrossRef]
- Matsukawa, Y.; Semba, S.; Kato, H.; Ito, A.; Yanagihara, K.; Yokozaki, H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006, 97, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Fujii, S.; Ochiai, A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008, 99, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional role of G9a histone methyltransferase in cancer. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Wei, S.; Hu, J.; Xiong, Z.; Lee, J.H. Upregulated expression of G9a is correlated with poor prognosis of gastric cancer patients. Medicne 2019, 98, e18212. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Huang, Y.; Zou, Y.; Chen, X.; Ma, X. Depletion of G9a gene induces cell apoptosis in human gastric carcinoma. Oncol. Rep. 2016, 35, 3041–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Ke, X.; Zhang, R.; Hou, J.; Dong, Z.; Wang, F.; Zhang, K.; Zhong, X.; Yang, L.; Cui, H. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 2019, 33, 14036–14050. [Google Scholar] [CrossRef] [Green Version]
- Estève, P.O.; Hang, G.C.; Smallwood, A.; Feehery, G.R.; Gangisetty, O.; Karpf, A.R.; Carey, M.F.; Pradhan, S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006, 20, 3089–3103. [Google Scholar] [CrossRef] [Green Version]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 2018, 10, 17. [Google Scholar] [CrossRef]
- Hu, L.; Zang, M.; Wang, H.-X.; Zhang, B.-G.; Wang, Z.-Q.; Fan, Z.-Y.; Wu, H.; Li, J.-F.; Su, L.-P.; Yan, M.; et al. G9A promotes gastric cancer metastasis by upregulating ITGB3 in a SET domain-independent manner. Cell Death Dis. 2018, 9, 278. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Schizas, D.; Mastoraki, A.; Naar, L.; Tsilimigras, D.I.; Katsaros, I.; Fragkiadaki, V.; Karachaliou, G.S.; Arkadopoulos, N.; Liakakos, T.; Moris, D. Histone Deacetylases (HDACs) in gastric cancer: An update of their emerging prognostic and therapeutic role. Curr. Med. Chem. 2019, 26. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amnekar, R.V.; Khan, S.A.; Rashid, M.; Khade, B.; Thorat, R.; Gera, P.; Shrikhande, S.V.; Smoot, D.T.; Ashktorab, H.; Gupta, S. Histone deacetylase inhibitor pre-treatment enhances the efficacy of DNA-interacting chemotherapeutic drugs in gastric cancer. World J. Gastroenterol. 2020, 26, 598–613. [Google Scholar] [CrossRef] [PubMed]
- Ono, S.; Oue, N.; Kuniyasu, H.; Suzuki, T.; Ito, R.; Matsusaki, K.; Ishikawa, T.; Tahara, E.; Yasui, W. Acetylated histone H4 is reduced in human gastric adenomas and carcinomas. J. Exp. Clin. Cancer Res. 2002, 21, 377–382. [Google Scholar]
- Sudo, T.; Mimori, K.; Nishida, N.; Kogo, R.; Iwaya, T.; Tanaka, F.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 2011, 26, 777–782. [Google Scholar]
- Mutze, K.; Langer, R.; Becker, K.; Ott, K.; Novotny, A.; Luber, B.; Hapfelmeier, A.; Göttlicher, M.; Höfler, H.; Keller, G. Histone deacetylase (HDAC) 1 and 2 expression and chemotherapy in gastric cancer. Ann. Surg. Oncol. 2010, 17, 3336–3343. [Google Scholar] [CrossRef]
- Noguchi, A.; Kikuchi, K.; Zheng, H.; Takahashi, H.; Miyagi, Y.; Aoki, I.; Takano, Y. SIRT1 expression is associated with a poor prognosis, whereas DBC1 is associated with favorable outcomes in gastric cancer. Cancer Med. 2014, 3, 1553–1561. [Google Scholar] [CrossRef]
- Lei, Z.; Tan, I.B.; Das, K.; Deng, N.; Zouridis, H.; Pattison, S.; Chua, C.; Feng, Z.; Guan, Y.K.; Ooi, C.H.; et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 2013, 145, 554–565. [Google Scholar] [CrossRef]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2013, 91, 49–58. [Google Scholar] [CrossRef]
- Niwa, T.; Toyoda, T.; Tsukamoto, T.; Mori, A.; Tatematsu, M.; Ushijima, T. Prevention of helicobacter pylori-induced gastric cancers in gerbils by a DNA demethylating agent. Cancer Prev. Res. 2013, 6, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Mok, M.T.S.; Li, M.S.M.; Kang, W.; Wang, H.; Chan, A.W.; Chou, J.L.; Chen, J.; Ng, E.K.W.; To, K.F.; et al. Epigenetic silencing of GDF1 disrupts SMAD signaling to reinforce gastric cancer development. Oncogene 2016, 35, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.Y.; Kim, G.Y.; Kim, C.G.; Kim, W.J.; Kang, H.S.; Choi, Y.H. Anti-invasive effects of decitabine, a DNA methyltransferase inhibitor, through tightening of tight junctions and inhibition of matrix metalloproteinase activities in AGS human gastric carcinoma cells. Oncol. Rep. 2012, 28, 1043–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zouridis, H.; Deng, N.; Ivanova, T.; Zhu, Y.; Wong, B.; Huang, D.; Wu, Y.H.; Wu, Y.; Tan, I.B.; Liem, N.; et al. Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci. Transl. Med. 2012, 4, 156ra140. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Rosas, F.; Hernández-Oliveras, A.; Flores-Peredo, L.; Rodríguez, G.; Zarain-Herzberg, Á.; Caba, M.; Santiago-Garcia, J. Histone deacetylase inhibitors induce the expression of tumor suppressor genes per1 and per2 in human gastric cancer cells. Oncol. Lett. 2018, 16, 1981–1990. [Google Scholar] [PubMed]
- Sun, J.; Piao, J.; Li, N.; Yang, Y.; Kim, K.Y.; Lin, Z. Valproic acid targets HDAC1/2 and HDAC1/PTEN/Akt signalling to inhibit cell proliferation via the induction of autophagy in gastric cancer. FEBS J. 2019, 287, 2118–2133. [Google Scholar] [CrossRef]
- Dong, J.; Zheng, N.; Wang, X.; Tang, C.; Yan, P.; Zhou, H.-B.; Huang, J. A novel HDAC6 inhibitor exerts an anti-cancer effect by triggering cell cycle arrest and apoptosis in gastric cancer. Eur. J. Pharmacol. 2018, 828, 67–79. [Google Scholar] [CrossRef]
- Fattahi, S.; Golpour, M.; Amjadi-Moheb, F.; Sharifi-Pasandi, M.; Khodadadi, P.; Pilehchian-Langroudi, M.; Ashrafi, G.H.; Akhavan-Niaki, H. DNA methyltransferases and gastric cancer: Insight into targeted therapy. Epigenomics 2018, 10, 1477–1497. [Google Scholar] [CrossRef] [Green Version]
- Xiong, K.; Zhang, H.; Du, Y.; Tian, J.; Ding, S. Identification of HDAC9 as a viable therapeutic target for the treatment of gastric cancer. Exp. Mol. Med. 2019, 51, 100. [Google Scholar] [CrossRef] [Green Version]
- Schneider, B.J.; Shah, M.A.; Klute, K.; Ocean, A.; Popa, E.; Altorki, N.; Lieberman, M.; Schreiner, A.; Yantiss, R.; Christos, P.J.; et al. Phase I study of epigenetic priming with azacitidine prior to standard neoadjuvant chemotherapy for patients with resectable gastric and esophageal adenocarcinoma: Evidence of tumor hypomethylation as an indicator of major histopathologic response. Clin. Cancer Res. 2017, 23, 2673–2680. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.; Ryu, M.H.; Na, Y.S.; Ryoo, B.Y.; Lee, C.W.; Kang, Y.K. Vorinostat in combination with capecitabine plus cisplatin as a first-line chemotherapy for patients with metastatic or unresectable gastric cancer: Phase II study and biomarker analysis. Br. J. Cancer 2016, 114, 1185–1190. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, H.-Q.; Liu, F. DNA Methyltransferase Inhibitors and their Therapeutic Potential. Curr. Top. Med. Chem. 2018, 18, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Ciesielski, O.; Balcerczyk, A. The Methylation Status of the Epigenome: Its Emerging Role in the Regulation of Tumor Angiogenesis and Tumor Growth, and Potential for Drug Targeting. Cancers 2018, 10, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, H.; Yashiro, M.; Shinto, O.; Matsuzaki, T.; Hirakawa, K. DNA methyltransferase inhibitor 5-aza-CdR enhances the radiosensitivity of gastric cancer cells. Cancer Sci. 2009, 100, 181–188. [Google Scholar] [CrossRef]
- Regel, I.; Merkl, L.; Friedrich, T.; Burgermeister, E.; Zimmermann, W.; Einwächter, H.; Herrmann, K.; Langer, R.; Röcken, R.; Hofheinz, R.; et al. Pan-histone deacetylase inhibitor panobinostat sensitizes gastric cancer cells to anthracyclines via induction of CITED2. Gastroenterology 2012, 143, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Zopf, S.; Ocker, M.; Neureiter, D.; Alinger, B.; Gahr, S.; Neurath, M.F.; Di Fazio, P. Inhibition of DNA methyltransferase activity and expression by treatment with the pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma cell lines. BMC Cancer 2012, 12, 386. [Google Scholar] [CrossRef] [Green Version]
- Curry, E.; Green, I.; Chapman-Rothe, N.; Shamsaei, E.; Kandil, S.; Cherblanc, F.L.; Payne, L.; Bell, E.; Ganesh, T.; Srimongkolpithak, N.; et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenet. 2015, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Lou, H.; Pan, H.; Huang, Z.; Wang, Z.; Wang, D.I. Inhibition of G9a promoted 5-fluorouracil (5-FU) induced gastric cancer cell apoptosis: Via ROS/JNK signaling pathway in vitro and in vivo. RSC Adv. 2019, 9, 14662–14669. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef]
- Wozniak, R.J.; Klimecki, W.T.; Lau, S.S.; Feinstein, Y.; Futscher, B.W. 5-Aza-2′-deoxycytidine-mediated reductions in G9A histone methyltransferase and histone H3 K9 di-methylation levels are linked to tumor suppressor gene reactivation. Oncogene 2007, 26, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Barcena-Varela, M.; Caruso, S.; Llerena, S.; Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of Histone Methyltransferase G9a and DNA-Methyltransferase 1 for the Treatment of Experimental Hepatocellular Carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef]
- Yuan, L.W.; Yamashita, H.; Seto, Y. Glucose metabolism in gastric cancer: The cutting-edge. World J. Gastroenterol. 2016, 22, 2046–2059. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Iwasa, S.; Muro, K.; Satoh, T.; Hironaka, S.; Esaki, T.; Nishina, T.; Hara, H.; Machida, N.; Komatsu, Y.; et al. Phase 1 trial of avelumab (anti-PD-L1) in Japanese patients with advanced solid tumors, including dose expansion in patients with gastric or gastroesophageal junction cancer: The JAVELIN Solid Tumor JPN trial. Gastric Cancer 2019, 22, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, R.; Huang, K.K.; Qamra, A.; Kim, K.M.; Kim, S.T.; Kang, W.K.; Tan, A.L.K.; Lee, J.; Tan, P. Epigenomic promoter alterations predict for benefit from immune checkpoint inhibition in metastatic gastric cancer. Ann. Oncol. 2019, 30, 424–430. [Google Scholar] [CrossRef]
- Lv, D.; Xing, C.; Cao, L.; Zhuo, Y.; Wu, T.; Gao, N. PD-L1 gene promoter methylation represents a potential diagnostic marker in advanced gastric cancer. Oncol. Lett. 2020, 19, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Zhang, P.; Liu, W.; Zeng, X.; Ma, X.; Shi, L.; Wang, T.; Yin, Y.; Chang, W.; Zhang, P.; et al. HDAC is indispensable for IFN-gamma-induced B7-H1 expression in gastric cancer. Clin. Epigenet. 2018, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xiang, S.; Shen, J.; Xiao, M.; Zhao, Y.; Wu, X.; Du, F.; Ji, H.; Li, M.; Zhao, Q.; et al. Comprehensive understanding of B7 family in gastric cancer: Expression profile, association with clinicopathological parameters and downstream targets. Int. J. Biol. Sci. 2020, 16, 568–582. [Google Scholar] [CrossRef] [Green Version]
- Qamra, A.; Xing, M.; Padmanabhan, N.; Kwok, J.J.T.; Zhang, S.; Xu, C.; Leong, Y.S.; Lim, A.P.; Tang, Q.; Ooi, W.F.; et al. Epigenomic promoter alterations amplify gene isoform and immunogenic diversity in gastric adenocarcinoma. Cancer Discov. 2017, 7, 630–651. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.C.; Chen, C.W.; Yang, C.L.; Tsai, I.M.; Hou, Y.C.; Chen, C.J.; Shan, Y.S. Tumor-associated macrophages promote epigenetic silencing of gelsolin through DNA methyltransferase 1 in gastric cancer cells. Cancer Immunol. Res. 2017, 5, 885–897. [Google Scholar] [CrossRef] [Green Version]
- Loo Yau, H.; Ettayebi, I.; De Carvalho, D.D. The Cancer Epigenome: Exploiting Its Vulnerabilities for Immunotherapy. Trends Cell Biol. 2019, 29, 31–43. [Google Scholar] [CrossRef]
- Abdelfatah, E.; Yang, S.; Duncan, M.; Ahuja, N.; Taube, J.M. Gastric Adenocarcinomas and Associated Immune Stroma. Gut 2017, 66, 794–801. [Google Scholar]
- Zhu, T.; Hu, Z.; Wang, Z.; Ding, H.; Li, R.; Sun, J.; Wang, G. Epigenetically silenced PD-L1 confers drug resistance to anti-PD1 therapy in gastric cardia adenocarcinoma. Int. Immunopharmacol. 2020, 82, 106245. [Google Scholar] [CrossRef] [PubMed]
- Olino, K.; Park, T.; Ahuja, N. Exposing Hidden Targets: Combining epigenetic and immunotherapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]


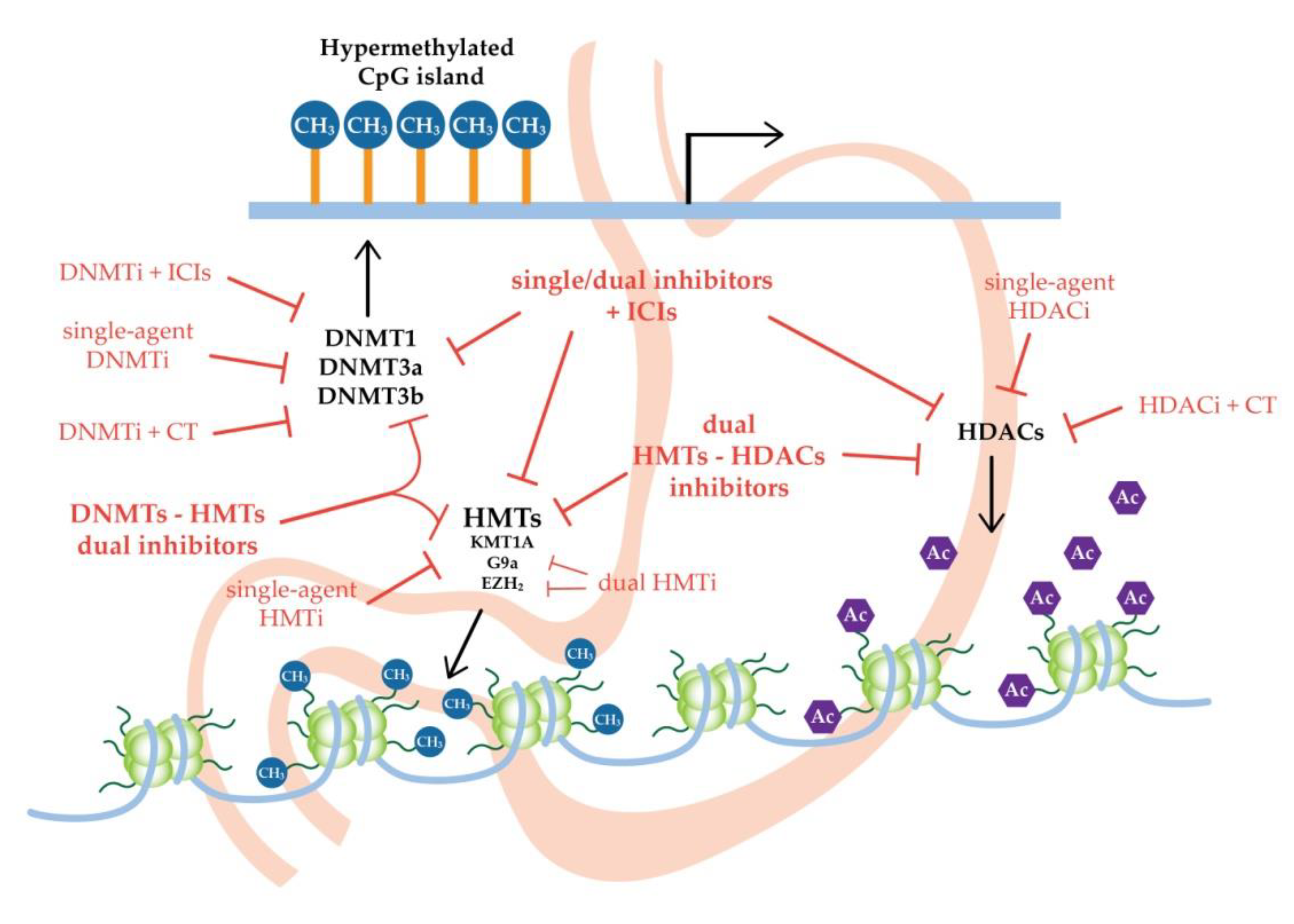{kind=link}
| Target | Role | Ref. |
|---|---|---|
| CDH1 | Cell–cell adhesion | [47] |
| MLH1, MLH2 | DNA repair | [58] |
| MGMT | DNA repair | [59] |
| DKK3 | Wnt signaling pathway regulation | [63] |
| RADSSF1A | Cell cycle regulation | [61] |
| HRAS | Component of RAS pathway | [62] |
| c-MYC | Transcription factor | [62] |
| CDKN2A | Cell cycle regulation | [64] |
| RUNX3 | Transcription factor | [73] |
| VEGF-c | Neo-angiogenesis related | [76] |
| GATA 4/5 | Gastrointestinal cell differentiation | [76] |
| Treatment Strategy | Epigenetic Target | Drug | Result | Model or Clinical Study Phase | Ref. |
|---|---|---|---|---|---|
| Single-agent | DNMTs | 5-azacitidine | Decreased GC incidence and decreased global hypermethylation in vivo | Mongolian gerbils | [133] |
| DNMTs | 5-azacitidine | Restoration of Gdf2-SMAD2/3 axis | MNU-treated mice | [134] | |
| DNMTs | DAC | Reduction of invasiveness of GC cells | GC cell lines | [135] | |
| DNMTs | DAC | Reduced cell growth in CIMP-positive cell lines | GC cell lines | [136] | |
| HDACs | TSA | Re-establishment of tumor suppressor gene expression | GC cell lines | [137] | |
| HDACs | VA | Inhibition of cell growth and apoptosis trigger | In vitro and in vivo models | [138] | |
| HDAC6 | TC24 | Cell cycle arrest and apoptosis, loss of mitochondrial membrane potential | GC cell lines | [139] | |
| Combination therapy, epigenetic priming | HDACs | VPA, TSA, SAHA, chemotherapy | Increase of DNA binding of cytotoxic agents and higher cytotoxic potential | GC cell lines | [126] |
| HMT G9a | G9a siRNA + 5-FU | Apoptosis trigger, synergism with 5-FU | GC cell lines | [140] | |
| HDAC9 | HDAC9 siRNA + cisplatin | Cell cycle arrest and apoptosis, synergism with cisplatin | In vitro and in vivo models | [141] | |
| DNMTs | 5-azacitidine prior to neoadjuvant chemotherapy | 67% overall response rate, 25% complete response | Phase I (NCT01386346) | [142] | |
| HDACs | SAHA + capecitabine, cisplatin | 42% objective response rate, increased adverse events | Phase II (NCT01045538) | [143] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canale, M.; Casadei-Gardini, A.; Ulivi, P.; Arechederra, M.; Berasain, C.; Lollini, P.-L.; Fernández-Barrena, M.G.; Avila, M.A. Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 5500. https://doi.org/10.3390/ijms21155500
Canale M, Casadei-Gardini A, Ulivi P, Arechederra M, Berasain C, Lollini P-L, Fernández-Barrena MG, Avila MA. Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities. International Journal of Molecular Sciences. 2020; 21(15):5500. https://doi.org/10.3390/ijms21155500
Chicago/Turabian StyleCanale, Matteo, Andrea Casadei-Gardini, Paola Ulivi, Maria Arechederra, Carmen Berasain, Pier-Luigi Lollini, Maite G. Fernández-Barrena, and Matías A. Avila. 2020. "Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities" International Journal of Molecular Sciences 21, no. 15: 5500. https://doi.org/10.3390/ijms21155500
APA StyleCanale, M., Casadei-Gardini, A., Ulivi, P., Arechederra, M., Berasain, C., Lollini, P. -L., Fernández-Barrena, M. G., & Avila, M. A. (2020). Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities. International Journal of Molecular Sciences, 21(15), 5500. https://doi.org/10.3390/ijms21155500