Persistent Human KIT Receptor Signaling Disposes Murine Placenta to Premature Differentiation Resulting in Severely Disrupted Placental Structure and Functionality

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
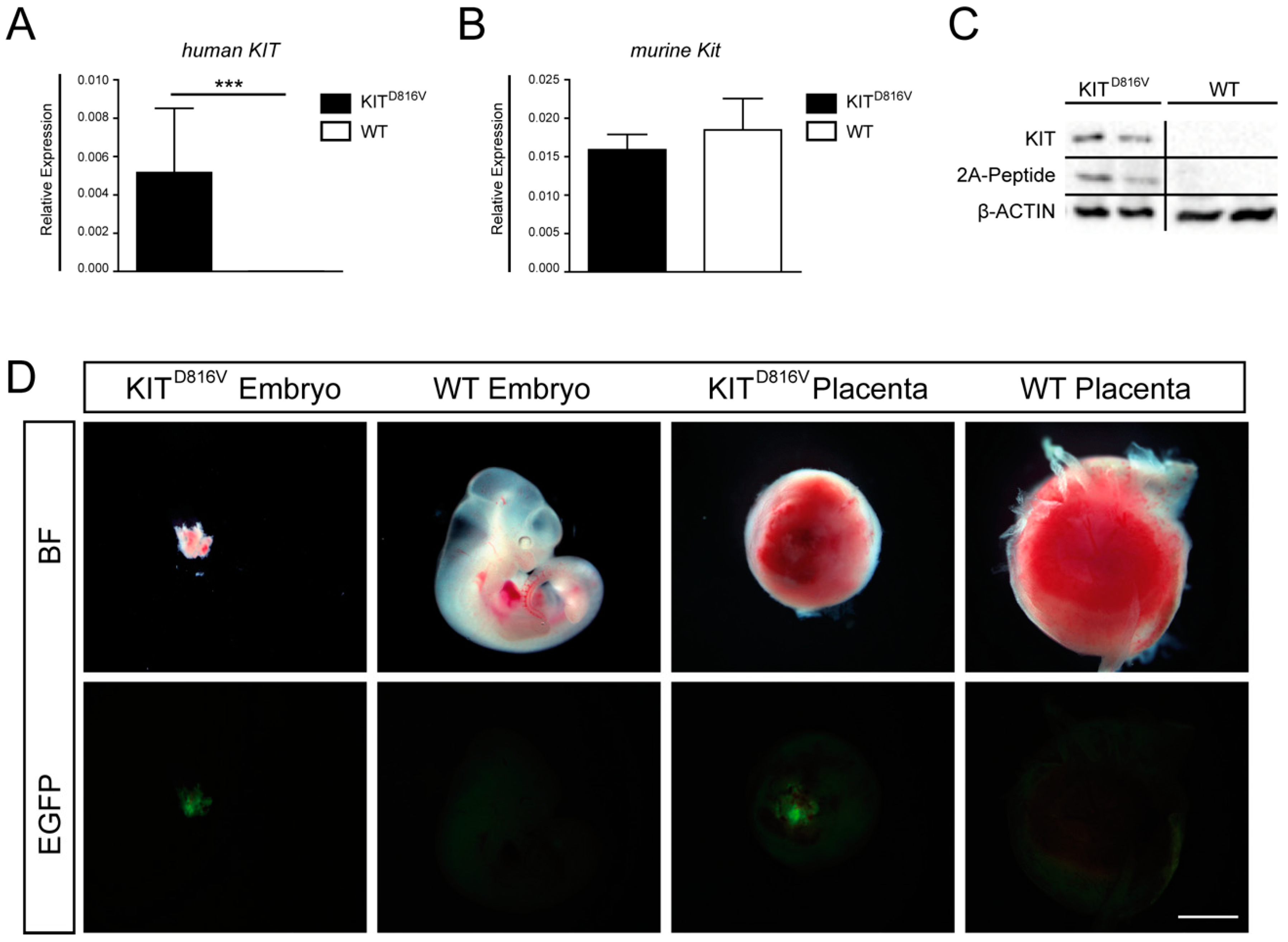
2.1. Embryos and Placentas Carrying KITD816V Mutation Suffer from Severe Growth Retardation
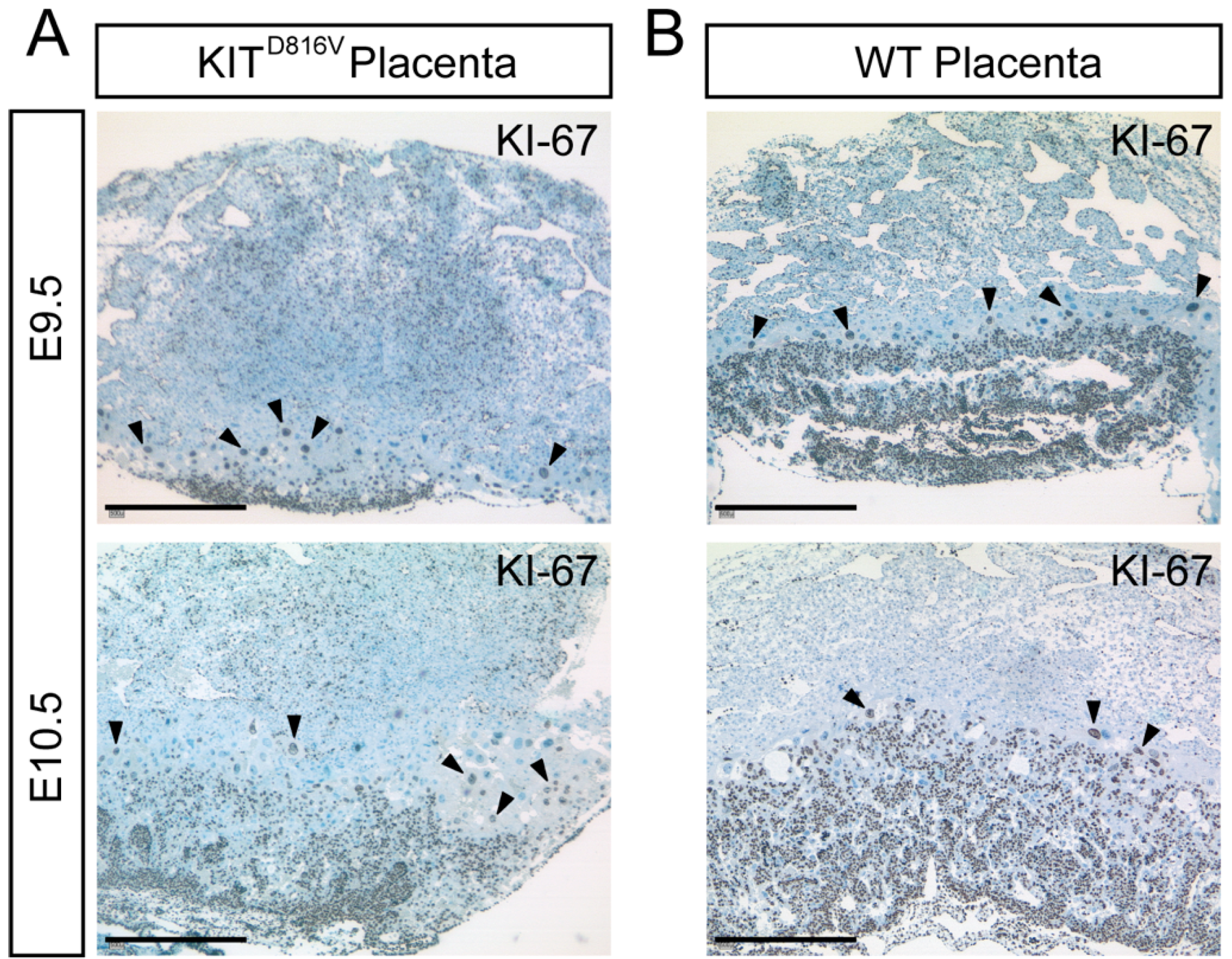
2.2. KITD816V Placentas Show Fewer Proliferating Cells
2.3. KITD816V Placentas Show Reduced Labyrinth Layer and Disrupted Formation of Vasculature
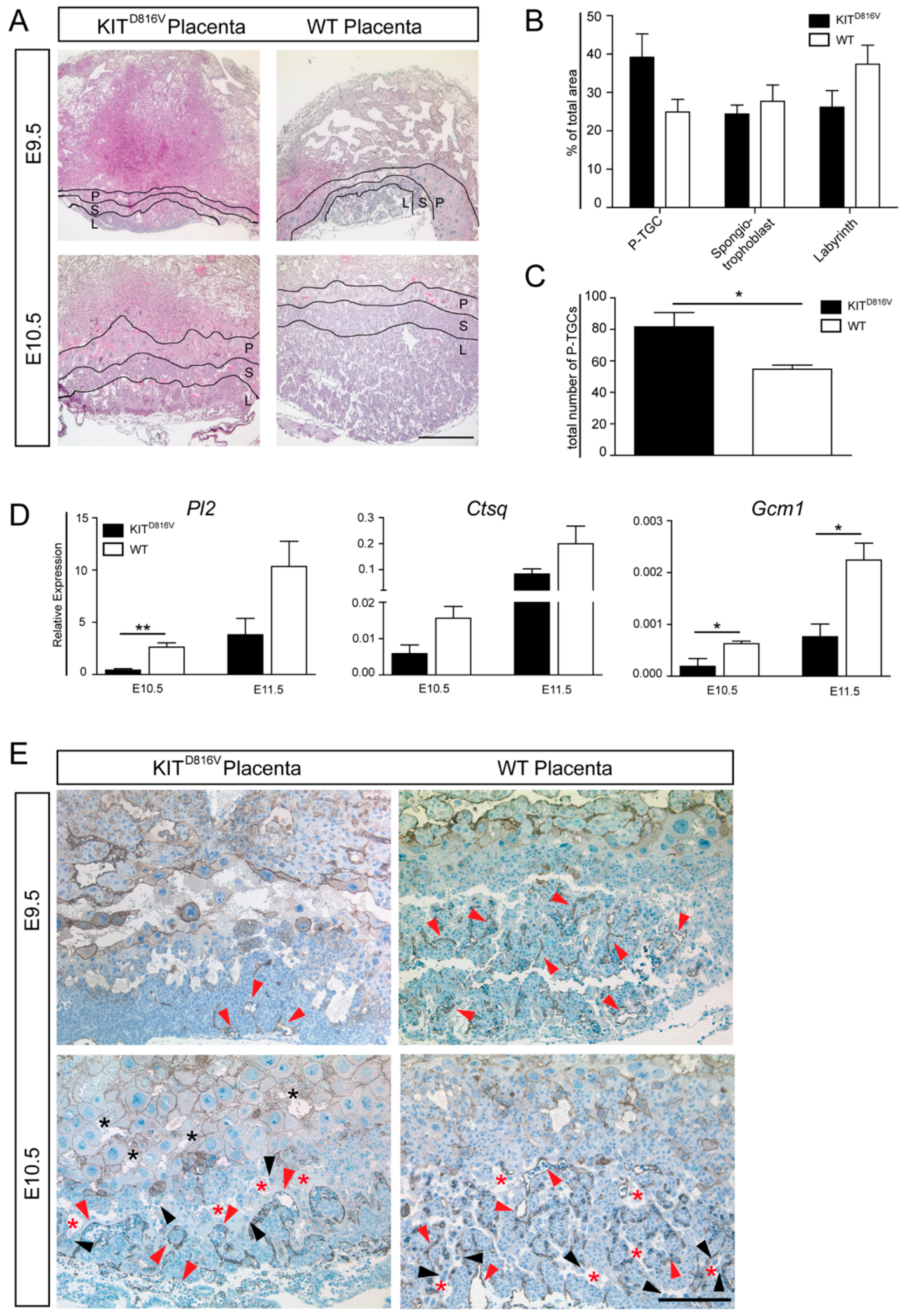
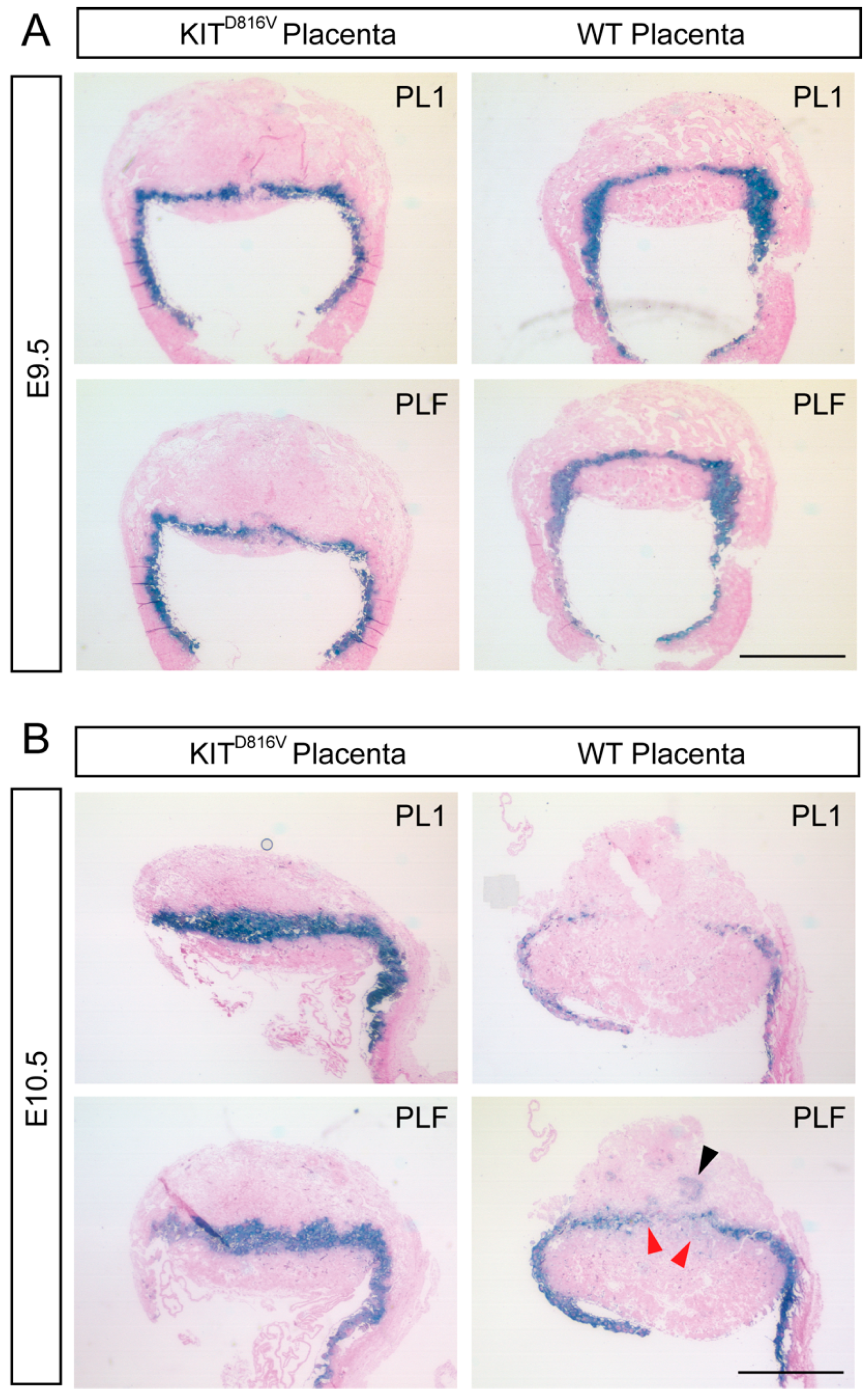
2.4. KITD816V Placentas Show Prominent Differentiation into P-TGCs
2.5. Spongiotrophoblast Cells and Glycogen Trophoblasts are Reduced in KITD816V Placentas
2.6. KITD816V-TSC Show Signs of Premature Differentiation
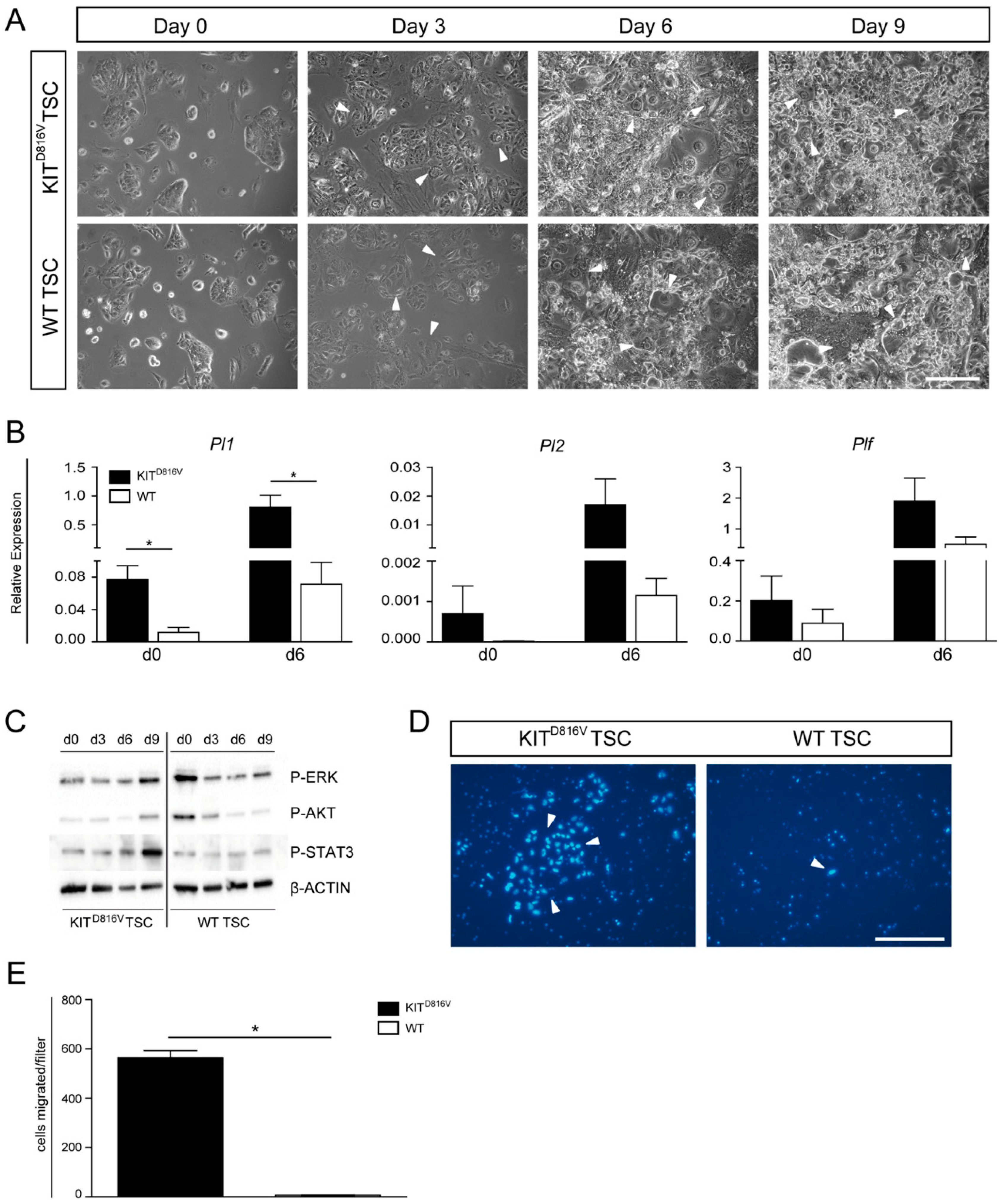
2.7. Signaling Cascades and Invasion Capability are Affected in KITD816V-TSC
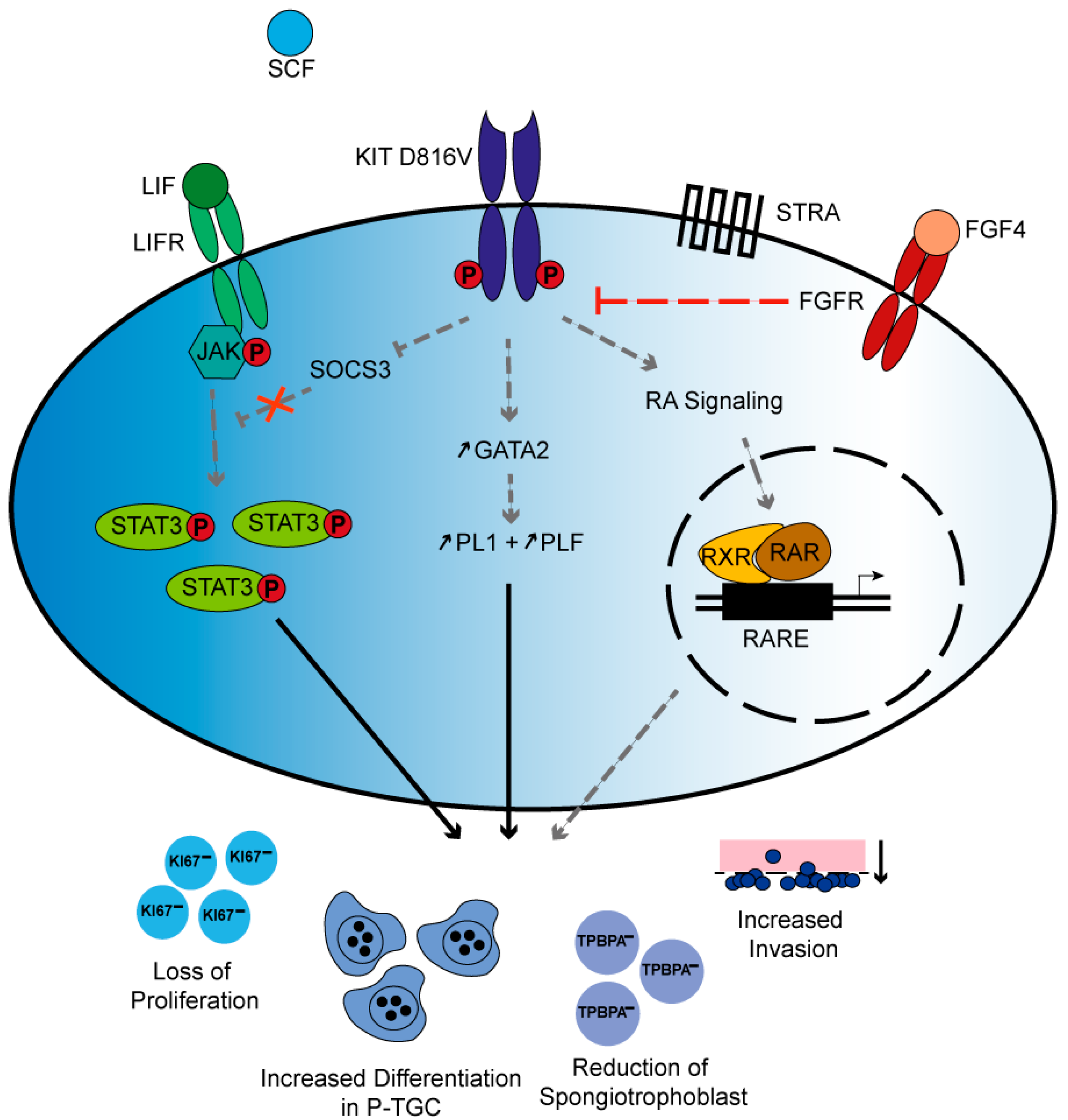
3. Discussion
4. Materials and Methods
4.1. Generation of Transgenic Animals
4.2. DNA Isolation and Genotyping PCR
4.3. RNA Analysis
4.4. Hematoxylin and Eosin Staining
4.5. Western Blotting
4.6. Immunohistochemical/Immunofluorescence Staining
4.7. In Situ Hybridization
4.8. Cell Culture
4.9. Flow Cytometric Analysis
4.10. Invasion Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein Kinase B |
| C-TGC | Canal TGC |
| CM | Conditioned Medium |
| ERK | Extracellular-Signal Regulated Kinase |
| FGF4 | Fibroblast Growth Factor 4 |
| FGFR | Fibroblast Growth Factor Receptor |
| GlyT | Glycogen Trophoblast |
| HSC | Hematopoietic Stem Cell |
| JAK | Janus Kinase |
| LIF | Leukemia Inhibitory Factor |
| MAPK | Mitogen-Activated Protein Kinase |
| P-TGC | Parietal TGC |
| RA | Retinoic Acid |
| RAR | Retinoic Acid Receptor |
| RXR | Retinoic X Receptor |
| S-TGC | Sinusoidal TGC |
| SOCS3 | Suppressor of Cytokine Signaling 3 |
| SpA | Spiral Artery |
| STAT3 | Signal Transducers and Activators of Transcription |
| STRA | Stimulated by Retinoic Acid |
| TGC | Trophoblast Giant Cell |
| TSC | Trophoblast Stem Celle |
| WT | Wildtype |
References
- Bazer, F.W.; Spencer, T.E.; Johnson, G.A.; Burghardt, R.C.; Wu, G. Comparative aspects of implantation. Reproduction 2009, 138, 195–209. [Google Scholar] [CrossRef] [Green Version]
- Cockburn, K.; Rossant, J. Making the blastocyst: Lessons from the mouse. J. Clin. Investig. 2010, 120, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Woods, L.; Perez-Garcia, V.; Hemberger, M. Regulation of Placental Development and Its Impact on Fetal Growth—New Insights From Mouse Models. Front. Endocrinol. 2018, 9, 570. [Google Scholar] [CrossRef] [Green Version]
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Chabot, B.; Stephenson, D.A.; Chapman, V.M.; Besmer, P.; Bernstein, A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 1988, 335, 88–89. [Google Scholar] [CrossRef]
- Copeland, N.G.; Gilbert, D.J.; Cho, B.C.; Donovan, P.J.; Jenkins, N.A.; Cosman, D.; Anderson, D.; Lyman, S.D.; Williams, D.E. Mast cell growth factor maps near the steel locus on mouse chromosome 10 and is deleted in a number of steel alleles. Cell 1990, 63, 175–183. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Y.-L.; Chen, B.-J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Linnekin, D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int. J. Biochem. Cell Biol. 1999, 31, 1053–1074. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Signaling by Kit protein-tyrosine kinase-The stem cell factor receptor. Biochem. Biophys. Res. Commun. 2005, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kurosaki, T. Regulation of lymphocyte fate by Ras/ERK signals. Cell Cycle 2008, 7, 3634–3640. [Google Scholar] [CrossRef]
- Horie, K.; Fujita, J.; Takakura, K.; Kanzaki, H.; Kaneko, Y.; Iwai, M.; Nakayama, H.; Mori, T. Expression of C-Kit Protein during Placental Development1. Biol. Reprod. 1992, 47, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motro, B.; van der Kooy, D.; Rossant, J.; Reith, A.; Bernstein, A. Contiguous patterns of c-kit and steel expression: Analysis of mutations at the W and Sl loci. Development 1991, 113, 1207–1221. [Google Scholar] [PubMed]
- Ottersbach, K.; Dzierzak, E. The Murine Placenta Contains Hematopoietic Stem Cells within the Vascular Labyrinth Region. Dev. Cell 2005, 8, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo Portilho, N.; Pelajo-Machado, M. Mechanism of hematopoiesis and vasculogenesis in mouse placenta. Placenta 2018, 69, 140–145. [Google Scholar] [CrossRef]
- Sasaki, T.; Mizuochi, C.; Horio, Y.; Nakao, K.; Akashi, K.; Sugiyama, D. Regulation of hematopoietic cell clusters in the placental niche through SCF/Kit signaling in embryonic mouse. Development 2010, 137, 3941 LP–3952. [Google Scholar] [CrossRef] [Green Version]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric mastocytosis is a clonal disease associated with D 816 v and other activating c-KIT mutations. J. Invest. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Kemmer, K.; Corless, C.L.; Fletcher, J.A.; McGreevey, L.; Haley, A.; Griffith, D.; Cummings, O.W.; Wait, C.; Town, A.; Heinrich, M.C. KIT mutations are common in testicular seminomas. Am. J. Pathol. 2004, 164, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Lasota, J.; Jasinski, M.; Sarlomo-Rikala, M.; Miettinen, M. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am. J. Pathol. 1999, 154, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Malaise, M.; Steinbach, D.; Corbacioglu, S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2009, 4, 77–82. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. KIT (CD117): A Review on Expression in Normal and Neoplastic Tissues, and Mutations and Their Clinicopathologic Correlation. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 205–220. [Google Scholar] [CrossRef]
- Sun, J.; Pedersen, M.; Rönnstrand, L. The D816V mutation of c-Kit circumvents a requirement for Src family kinases in c-Kit signal transduction. J. Biol. Chem. 2009, 284, 11039–11047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Q.; Frierson, H.F.; Krystal, G.W.; Moskaluk, C.A. Activating c-kit gene mutations in human germ cell tumors. Am. J. Pathol. 1999, 154, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, R.A.; Mintz, B. Prevention of genetic anemias in mice by microinjection of normal hematopoietic stem cells into the fetal placenta. Proc. Natl. Acad. Sci. USA 1979, 76, 5736–5740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broudy, V.C. Stem Cell Factor and Hematopoiesis. Blood 1997, 90, 1345–1364. [Google Scholar] [CrossRef] [Green Version]
- Lotinun, S.; Krishnamra, N. Disruption of c-Kit Signaling in KitW-sh/W-sh Growing Mice Increases Bone Turnover. Sci. Rep. 2016, 6, 31515. [Google Scholar] [CrossRef] [Green Version]
- Waskow, C.; Paul, S.; Haller, C.; Gassmann, M.; Rodewald, H.-R. Viable c-KitW/W Mutants Reveal Pivotal Role for c-Kit in the Maintenance of Lymphopoiesis. Immunity 2002, 17, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Haas, N.; Riedt, T.; Labbaf, Z.; Baßler, K.; Gergis, D.; Fröhlich, H.; Gütgemann, I.; Janzen, V.; Schorle, H. Kit transduced signals counteract erythroid maturation by MAPK-dependent modulation of erythropoietin signaling and apoptosis induction in mouse fetal liver. Cell Death Differ. 2015, 22, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Kreisel, F.; Cain, J.; Colson, A.; Tomasson, M.H. Neoplasia driven by mutant c-KIT is mediated by intracellular, not plasma membrane, receptor signaling. Mol. Cell. Biol. 2007, 27, 267–282. [Google Scholar] [CrossRef] [Green Version]
- Pelusi, N.; Kosanke, M.; Riedt, T.; Rösseler, C.; Seré, K.; Li, J.; Gütgemann, I.; Zenke, M.; Janzen, V.; Schorle, H. The spleen microenvironment influences disease transformation in a mouse model of KITD816V-dependent myeloproliferative neoplasm. Sci. Rep. 2017, 7, 41427. [Google Scholar] [CrossRef] [Green Version]
- Winking, H.; Gerdes, J.; Traut, W. Expression of the proliferation marker Ki-67 during early mouse development. Cytogenet. Genome Res. 2004, 105, 251–256. [Google Scholar] [CrossRef]
- Hu, D.; Cross, J.C. Development and function of trophoblast giant cells in the rodent placenta. Int. J. Dev. Biol. 2010, 54, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, M.; Mieda, M.; Ikeda, T.; Hamada, Y.; Nakamura, H.; Okamoto, H. R-spondin3 is required for mouse placental development. Dev. Biol. 2007, 301, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himeno, E.; Tanaka, S.; Kunath, T. Isolation and Manipulation of Mouse Trophoblast Stem Cells. Curr. Protoc. Stem Cell Biol. 2008, 7, 1E.4.1–1E.4.27. [Google Scholar] [CrossRef] [PubMed]
- Kuckenberg, P.; Buhl, S.; Woynecki, T.; van Fürden, B.; Tolkunova, E.; Seiffe, F.; Moser, M.; Tomilin, A.; Winterhager, E.; Schorle, H. The Transcription Factor TCFAP2C/AP-2γ Cooperates with CDX2 To Maintain Trophectoderm Formation. Mol. Cell. Biol. 2010, 30, 3310–3320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, I.C.; Anson-Cartwright, L.; Riley, P.; Reda, D.; Cross, J.C. The HAND1 Basic Helix-Loop-Helix Transcription Factor Regulates Trophoblast Differentiation via Multiple Mechanisms. Mol. Cell. Biol. 2000, 20, 530 LP–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeuner, A.; Francescangeli, F.; Signore, M.; Venneri, M.A.; Pedini, F.; Felli, N.; Pagliuca, A.; Conticello, C.; De Maria, R. The Notch2-Jagged1 interaction mediates stem cell factor signaling in erythropoiesis. Cell Death Differ. 2011, 18, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Simmons, D.G.; Fortier, A.L.; Cross, J.C. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev. Biol. 2007, 304, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Hemberger, M.; Hughes, M.; Cross, J.C. Trophoblast stem cells differentiate in vitro into invasive trophoblast giant cells. Dev. Biol. 2004, 271, 362–371. [Google Scholar] [CrossRef]
- Rai, A.; Cross, J.C. Development of the hemochorial maternal vascular spaces in the placenta through endothelial and vasculogenic mimicry. Dev. Biol. 2014, 387, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Muta, K.; Krantz, S.B.; Bondurant, M.C.; Dai, C.H. Stem cell factor retards differentiation of normal human erythroid progenitor cells while stimulating proliferation. Blood 1995, 86, 572–580. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.T.; Roth, M.E.; Groskopf, J.C.; Tsai, F.Y.; Orkin, S.H.; Grosveld, F.; Engel, J.D.; Linzer, D.I. GATA-2 and GATA-3 regulate trophoblast-specific gene expression in vivo. Development 1997, 124, 907. [Google Scholar] [PubMed]
- Ng, Y.K.; George, K.M.; Engel, J.D.; Linzer, D.I. GATA factor activity is required for the trophoblast-specific transcriptional regulation of the mouse placental lactogen I gene. Development 1994, 120, 3257. [Google Scholar] [PubMed]
- Latos, P.A.; Hemberger, M. From the stem of the placental tree: Trophoblast stem cells and their progeny. Development 2016, 143, 3650 LP–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, D.G.; Cross, J.C. Determinants of trophoblast lineage and cell subtype specification in the mouse placenta. Dev. Biol. 2005, 284, 12–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Kunath, T.; Hadjantonakis, A.-K.; Nagy, A.; Rossant, J. Promotion of Trophoblast Stem Cell Proliferation by FGF4. Science (80-) 1998, 282, 2072 LP–2075. [Google Scholar] [CrossRef]
- Yan, J.; Tanaka, S.; Oda, M.; Makino, T.; Ohgane, J.; Shiota, K. Retinoic acid promotes differentiation of trophoblast stem cells to a giant cell fate. Dev. Biol. 2001, 235, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busada, J.T.; Chappell, V.A.; Niedenberger, B.A.; Kaye, E.P.; Keiper, B.D.; Hogarth, C.A.; Geyer, C.B. Retinoic acid regulates Kit translation during spermatogonial differentiation in the mouse. Dev. Biol. 2015, 397, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Gely-Pernot, A.; Raverdeau, M.; Teletin, M.; Vernet, N.; Féret, B.; Klopfenstein, M.; Dennefeld, C.; Davidson, I.; Benoit, G.; Mark, M.; et al. Retinoic Acid Receptors Control Spermatogonia Cell-Fate and Induce Expression of the SALL4A Transcription Factor. PLoS Genet. 2015, 11, e1005501. [Google Scholar] [CrossRef]
- Koli, S.; Mukherjee, A.; Reddy, K.V.R. Retinoic acid triggers c-kit gene expression in spermatogonial stem cells through an enhanceosome constituted between transcription factor binding sites for retinoic acid response element (RARE), spleen focus forming virus proviral integration onco. Reprod. Fertil. Dev. 2017, 29, 521–543. [Google Scholar] [CrossRef]
- Suman, P.; Malhotra, S.S.; Gupta, S.K. LIF-STAT signaling and trophoblast biology. JAK-STAT 2013, 2, e25155. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Takahashi, M.; Carpino, N.; Jou, S.-T.; Chao, J.-R.; Tanaka, S.; Shigeyoshi, Y.; Parganas, E.; Ihle, J.N. Leukemia Inhibitory Factor Regulates Trophoblast Giant Cell Differentiation via Janus Kinase 1-Signal Transducer and Activator of Transcription 3-Suppressor of Cytokine Signaling 3 Pathway. Mol. Endocrinol. 2008, 22, 1673–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Carpino, N.; Cross, J.C.; Torres, M.; Parganas, E.; Ihle, J.N. SOCS3: An essential regulator of LIF receptor signaling in trophoblast giant cell differentiation. EMBO J. 2003, 22, 372–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S. Regulation of Metastases by Signal Transducer and Activator of Transcription 3 Signaling Pathway: Clinical Implications. Clin. Cancer Res. 2007, 13, 1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwenk, F.; Baron, U.; Rajewsky, K. A cre -transgenic mouse strain for the ubiquitous deletion of loxP -flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995, 23, 5080–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaiser, F.; Hartweg, J.; Jansky, S.; Pelusi, N.; Kubaczka, C.; Sharma, N.; Nitsche, D.; Langkabel, J.; Schorle, H. Persistent Human KIT Receptor Signaling Disposes Murine Placenta to Premature Differentiation Resulting in Severely Disrupted Placental Structure and Functionality. Int. J. Mol. Sci. 2020, 21, 5503. https://doi.org/10.3390/ijms21155503
Kaiser F, Hartweg J, Jansky S, Pelusi N, Kubaczka C, Sharma N, Nitsche D, Langkabel J, Schorle H. Persistent Human KIT Receptor Signaling Disposes Murine Placenta to Premature Differentiation Resulting in Severely Disrupted Placental Structure and Functionality. International Journal of Molecular Sciences. 2020; 21(15):5503. https://doi.org/10.3390/ijms21155503
Chicago/Turabian StyleKaiser, Franziska, Julia Hartweg, Selina Jansky, Natalie Pelusi, Caroline Kubaczka, Neha Sharma, Dominik Nitsche, Jan Langkabel, and Hubert Schorle. 2020. "Persistent Human KIT Receptor Signaling Disposes Murine Placenta to Premature Differentiation Resulting in Severely Disrupted Placental Structure and Functionality" International Journal of Molecular Sciences 21, no. 15: 5503. https://doi.org/10.3390/ijms21155503
APA StyleKaiser, F., Hartweg, J., Jansky, S., Pelusi, N., Kubaczka, C., Sharma, N., Nitsche, D., Langkabel, J., & Schorle, H. (2020). Persistent Human KIT Receptor Signaling Disposes Murine Placenta to Premature Differentiation Resulting in Severely Disrupted Placental Structure and Functionality. International Journal of Molecular Sciences, 21(15), 5503. https://doi.org/10.3390/ijms21155503