Comparative Transcriptomic Analysis of the Development of Sepal Morphology in Tomato (Solanum Lycopersicum L.)


 and
and
Abstract
:1. Introduction
2. Results
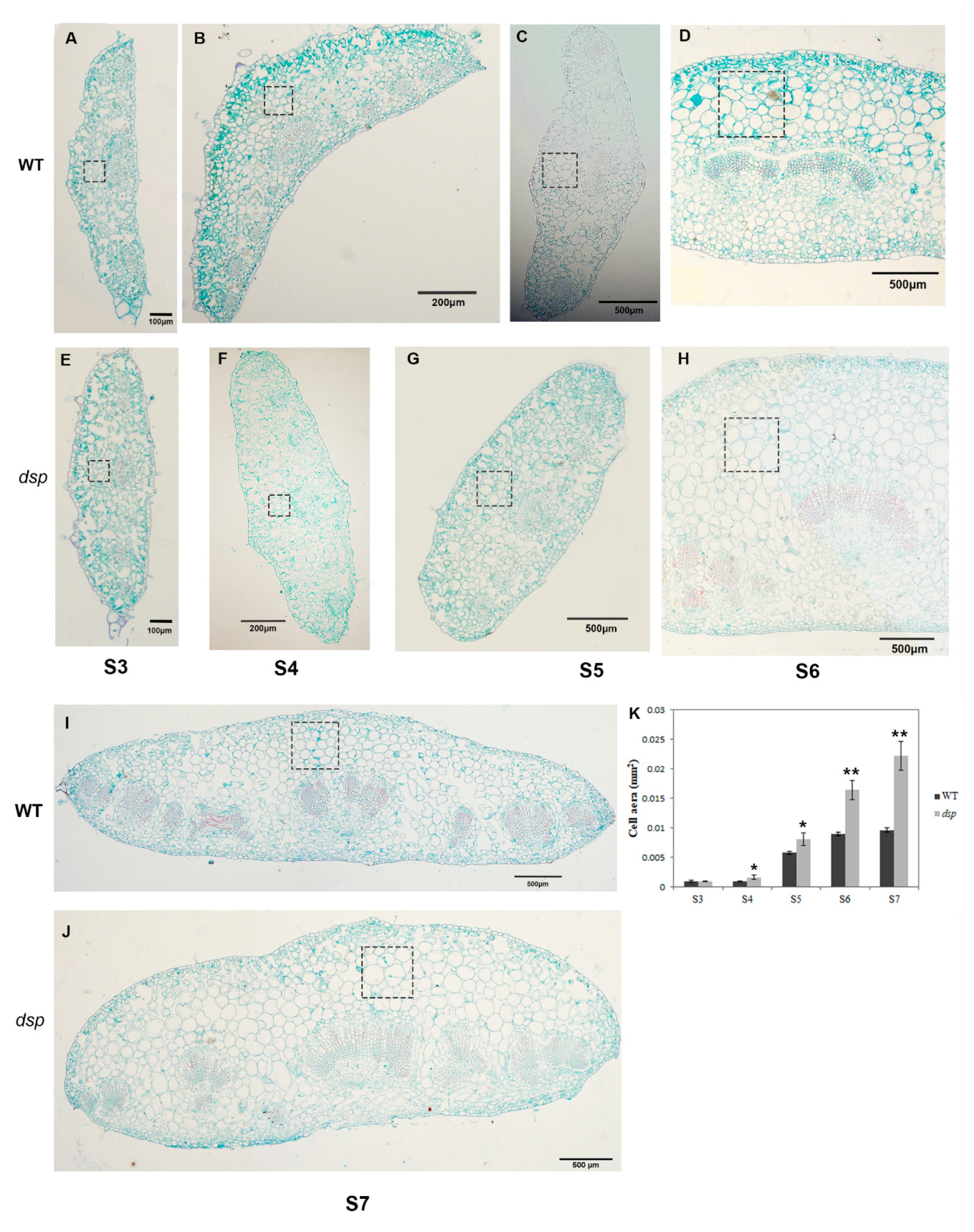
2.1. Phenotypic Development Features and Diversity of Tomato Sepals
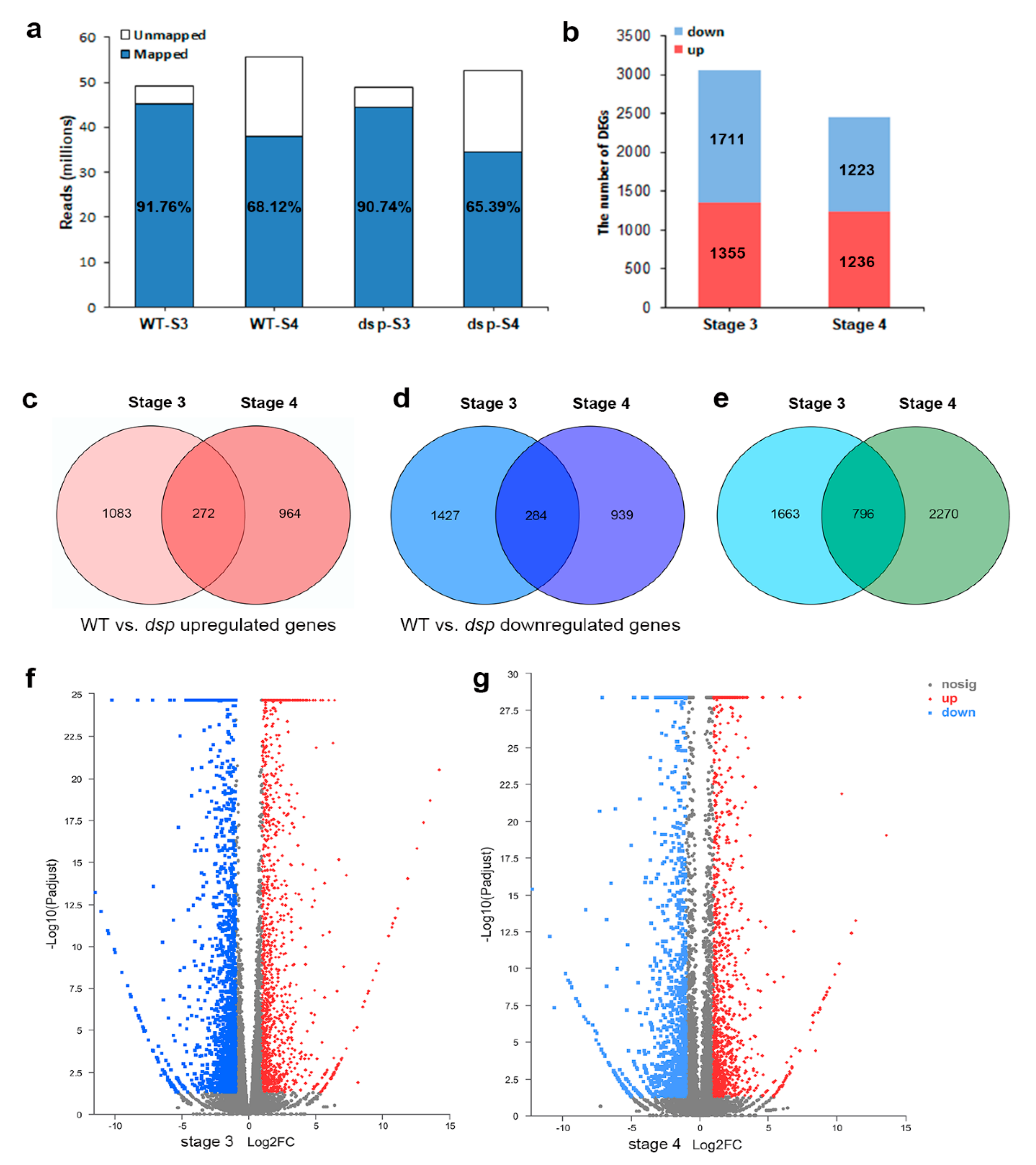
2.2. Identification of Differentially Expressed Genes in Sepals from WT and dsp Mutant Plants
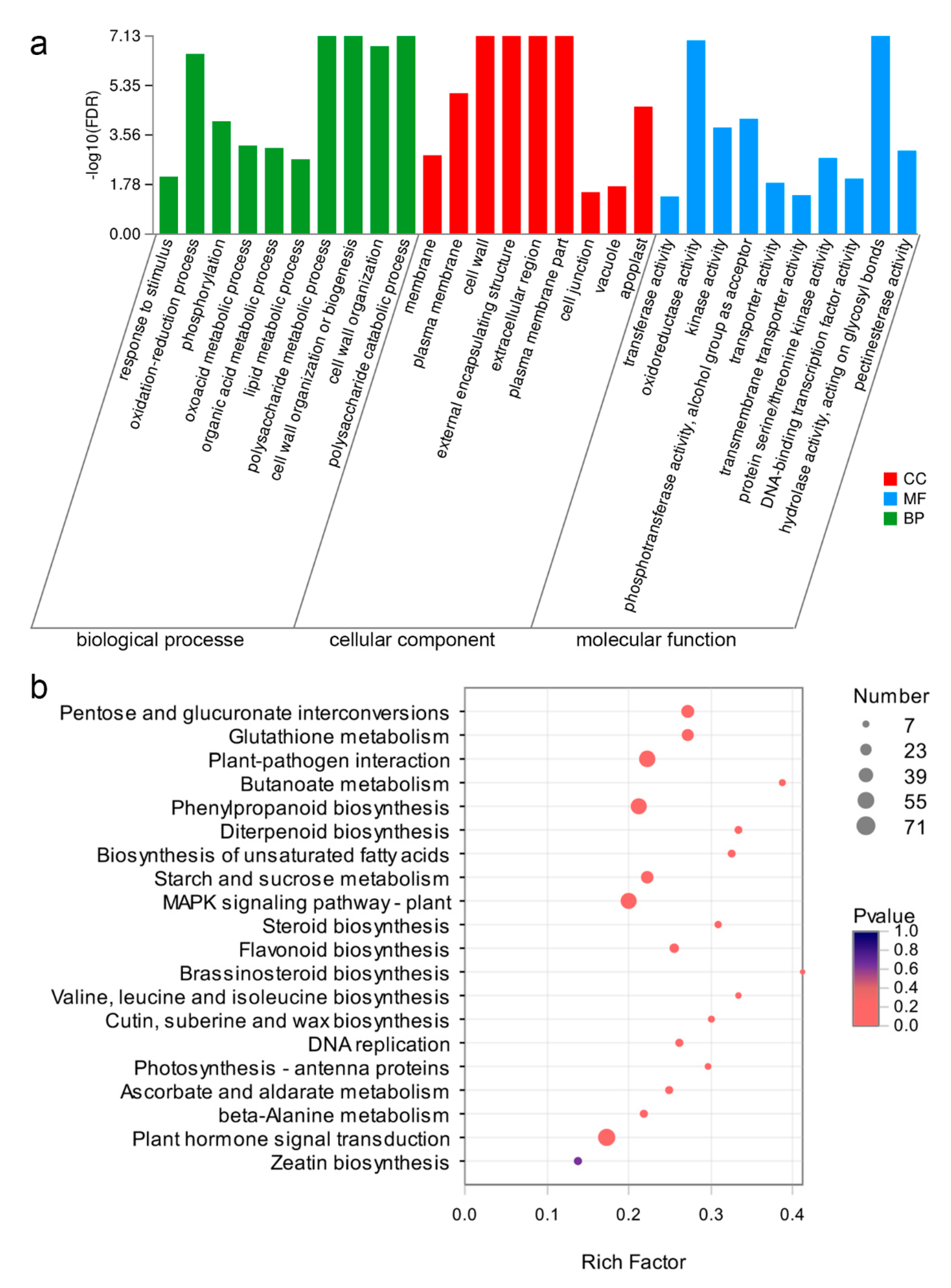
2.3. Annotation of DEGs in Sepals from WT and dsp Mutant Plants
2.4. Coexpression Pattern of DEGs in Sepals from WT and dsp Mutant Plants
2.5. Regulation of Cell Expansion by dsp to Control Sepal Morphology
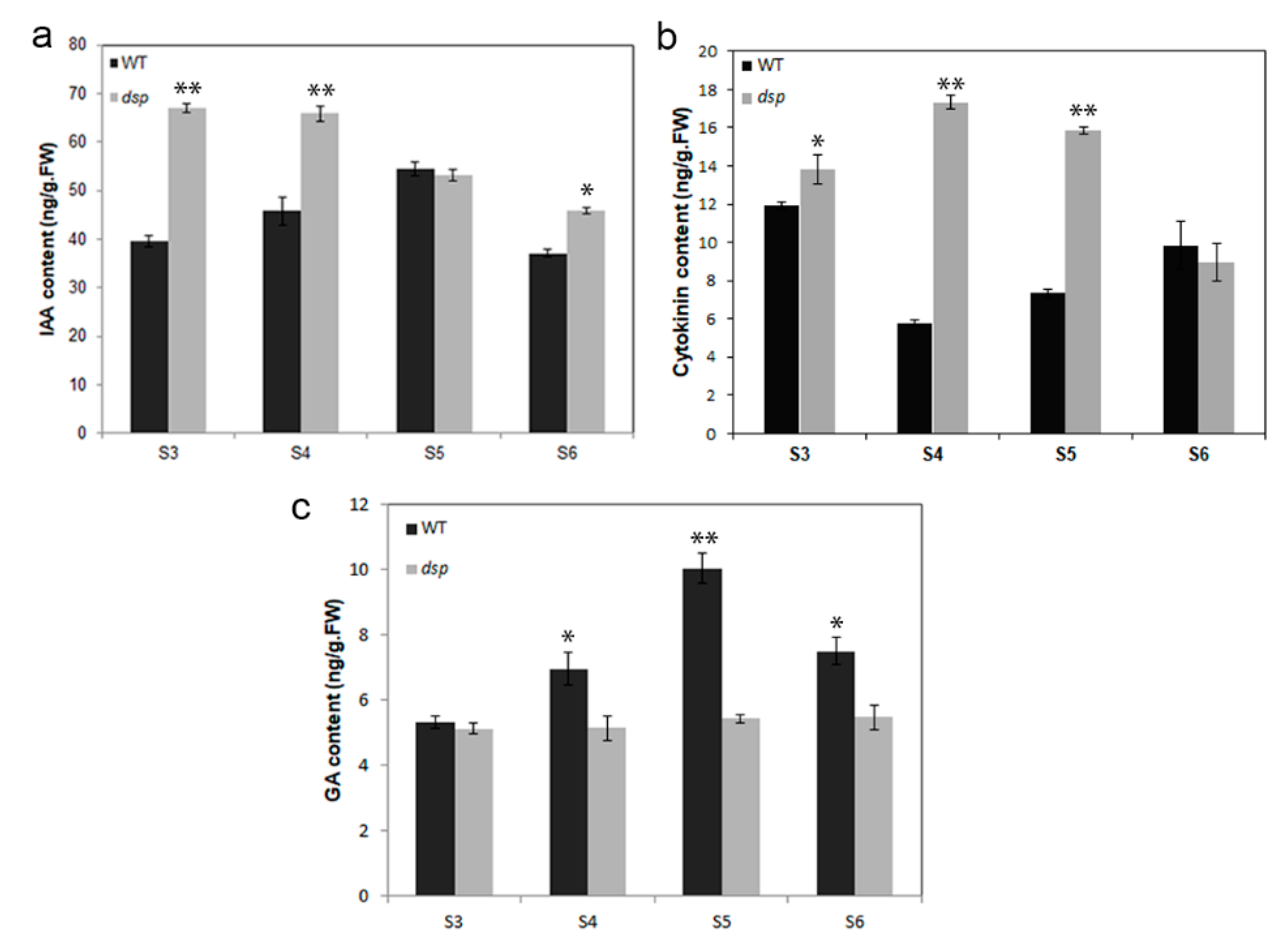
2.6. Effects of dsp on Auxin, Cytokinin, and Gibberellin Levels in Sepals
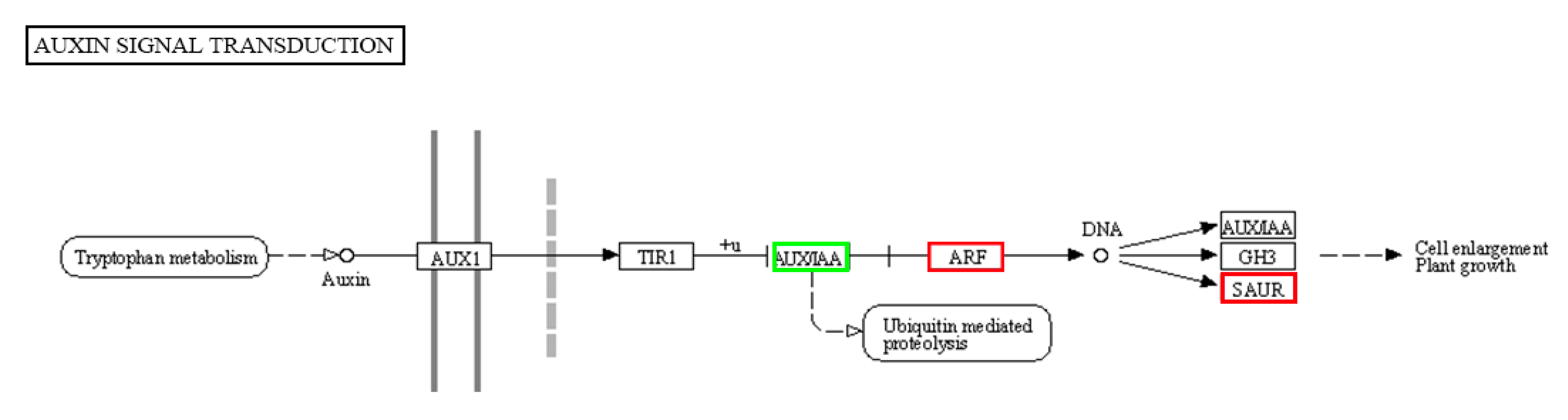
2.7. Possible Function of 17 Auxin-Responsive Genes as Regulators in Downward Sepal Formation in the Tomato
3. Discussion
4. Materials and Methods
4.1. Plant Materials
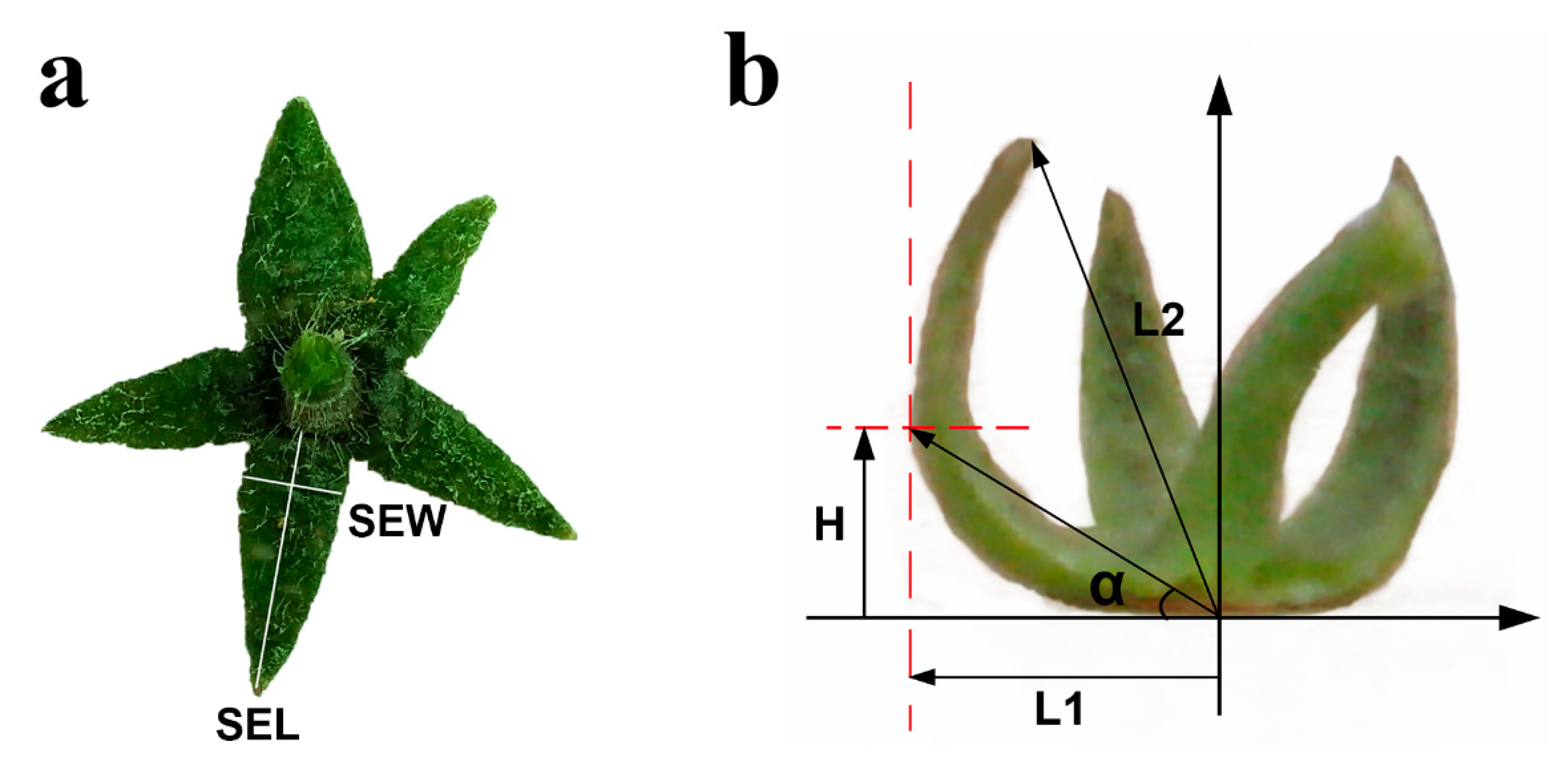
4.2. Measurement of Sepal Morphology at Different Developmental Stages
- (1)
- Sepal Upturned Degree (SEUD, °) = 90° + α = 90° + arctan (H/L1), degree of upwarp or down-wrap of sepal. Put a whole calyx on a L-square ruler, then keep the calyx base level on one side of L-square ruler and the sepal to be measured leaning against another side of L-square ruler. H represents the vertical distance between the furthest point from sepal to stalk in the horizontal line and the base line, L1 is the longest distance from sepal to stalk (Figure 9).
- (2)
- Sepal Rolling Index (SERI, %) = (SEL − L2)/SEL × 100%, SEL (Sepal Length, mm) is the length of sepal in flat condition, L2 is the distance from apex to base point (Figure 9).
4.3. DGE (Differentially Gene Expression) Library Construction and Sequencing
4.4. Bioinformatics Analysis of DGE Data
4.5. Paraffin Sectioning and Electron Microscopy Experiment
4.6. Extraction, Purification and Quantification of the Phytohormones in Sepals
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhong, F.L.; Zhou, X.Z.; Lin, Y.W.; Chen, X.; Xu, R.; Wang, S.B.; Lin, Y.Z.; Pang, J.; Wu, S. Sepal Morphology Affects Flower and Fruit Development in Luffa Cylindrica. Fresenius. Environ. Bull. 2018, 27, 4006–4013. [Google Scholar]
- Li, N.; Huang, B.; Tang, N.; Jian, W.; Zou, J.; Chen, J.; Cao, H.; Habib, S.; Dong, X.; Wei, W.; et al. The MADS-Box Gene SlMBP21 Regulates Sepal Size Mediated by Ethylene and Auxin in Tomato. Plant Cell. Physiol. 2017, 58, 2241–2256. [Google Scholar] [CrossRef] [PubMed]
- Vemmos, S.N.; Goldwin, G.K. The Photosynthetic Activity of Cox Orange Pippin Apple Flowers in Relation to Fruit Setting. Ann. Bot. 1994, 73, 385–391. [Google Scholar] [CrossRef]
- Hong, L.L.; Dumond, M.; Tsugawa, S.; Sapala, A.; Routier-Kierzkowska, A.L.; Zhou, Y.; Chen, C.; Kiss, A.; Zhu, M.Y.; Hamant, O.; et al. Variable Cell Growth Yields Reproducible Organ Development through Spatiotemporal Averaging. Dev. Cell. 2016, 38, 15–32. [Google Scholar] [CrossRef] [Green Version]
- Tauriello, G.; Meyer, H.M.; Smith, R.S.; Koumoutsakos, P.; Roeder, A.H.K. Variability and Constancy in Cellular Growth of Arabidopsis Sepals. Plant Physiol. 2015, 169, 2342–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santner, A.; Calderon-Villalobos, L.I.A.; Estelle, M. Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 2009, 5, 301–307. [Google Scholar] [CrossRef]
- Wang, Y.H.; Irving, H.R. Developing a model of plant hormone interactions. Plant Signal. Behav. 2011, 6, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Varaud, E.; Brioudes, F.; Szécsi, J.; Leroux, J.; Brown, S.; Perrot-Rechenmann, C.; Bendahmane, M. AUXIN RESPONSE FACTOR8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell. 2011, 23, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Hua, C.; Shen, L.; Yu, H. New insights into gibberellin signaling in regulating flowering in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Danisman, S.; Van Dijk, A.D.J.; Bimbo, A.; Van der Wal, F.; Hennig, L.; De Folter, S.; Angenent, G.C.; Immink, R.G.H. Analysis of functional redundancies within the Arabidopsis TCP transcription factor family. J. Exp. Bot. 2013, 64, 5673–5685. [Google Scholar] [CrossRef] [Green Version]
- Koyama, T.; Mitsuda, N.; Seki, M.; Shinozaki, K.; Ohme-Takagi, M. TCP Transcription Factors Regulate the Activities of ASYMMETRIC LEAVES1 and miR164, as Well as the Auxin Response, during Differentiation of Leaves in Arabidopsis. Plant Cell. 2010, 22, 3574–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, A.; King, S.; Jack, T. miR319a targeting of TCP4 is critical for petal growth and development in Arabidopsis. Proc. Natl. Acad. Sci. USA 2009, 106, 22534–22539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.Y.; Zhao, P.M.; Cheng, H.Q.; Han, L.B.; Wu, X.M.; Gao, P.; Wang, H.Y.; Yang, C.L.; Zhong, N.Q.; Zuo, J.R.; et al. The Cotton Transcription Factor TCP14 Functions in Auxin-Mediated Epidermal Cell Differentiation and Elongation. Plant Physiol. 2013, 162, 1669–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrebalov, J.; Ruezinsky, D.; Padmanabhan, V.; White, R.; Medrano, D.; Drake, R.; Schuch, W.; Giovannoni, J. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor (Rin) locus. Science 2002, 296, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Giménez, E.; Pineda, B.; Capel, J.; Teresa Antón, M.; Atarés, A.; Pérez-Martin, F.; Garcia-Sogo, B.; Angosto, T.; Moreno, V.; Lozano, R. Functional Analysis of the Arlequin Mutant Corroborates the Essential Role of the ARLEQUIN/TAGL1 Gene during Reproductive Development of Tomato. PLoS ONE 2010, 5, e14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.L.; Hu, Z.L.; Zhu, Z.G.; Dong, T.T.; Zhao, Z.P.; Cui, B.L.; Chen, G.P. Overexpression of a novel MADS-box gene SlFYFL delays senescence, fruit ripening and abscission in tomato. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Wang, D.; Qin, Z.; Zhang, D.; Yin, L.; Wu, L.; Colasanti, J.; Li, A.; Mao, L. The SEPALLATA MADS-box protein SLMBP21 forms protein complexes with JOINTLESS and MACROCALYX as a transcription activator for development of the tomato flower abscission zone. Plant J. 2014, 77, 284–296. [Google Scholar] [CrossRef]
- Roldan, M.V.G.; Perilleux, C.; Morin, H.; Huerga-Fernandez, S.; Latrasse, D.; Benhamed, M.; Bendahmane, A. Natural and induced loss of function mutations in SlMBP21 MADS-box gene led to jointless-2 phenotype in tomato. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Zhang, J.l.; Hu, Z.l.; Wang, Y.s.; Yu, X.h.; Liao, C.g.; Zhu, M.k.; Chen, G.p. Suppression of a tomato SEPALLATA MADS-box gene, SlCMB1, generates altered inflorescence architecture and enlarged sepals. Plant Sci. 2018, 272, 75–87. [Google Scholar] [CrossRef]
- Eveland, A.L.; Satoh-Nagasawa, N.; Goldshmidt, A.; Meyer, S.; Beatty, M.; Sakai, H.; Ware, D.; Jackson, D. Digital gene expression signatures for maize development. Plant Physiol. 2010, 154, 1024–1039. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, Z.G.; Tu, Y.; Cheng, W.J.; Yang, Y.W. Tomato (Solarium lycopersicum) SIIPT4, encoding an isopentenyltransferase, is involved in leaf senescence and lycopene biosynthesis during fruit ripening. BMC Plant Biol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Ding, J.; Lin, S.S.; Liu, D.C.; Gu, T.T.; Wu, H.; Trigiano, R.N.; McAvoy, R.; Huang, J.L.; Li, Y. Evolution and roles of cytokinin genes in angiosperms 2: Do ancient CKXs play housekeeping roles while non-ancient CKXs play regulatory roles? Hortic. Res.-Engl. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Veach, Y.K.; Martin, R.C.; Mok, D.W.; Malbeck, J.; Vankova, R.; Mok, M.C. O-glucosylation of cis-zeatin in maize. Characterization of genes, enzymes, and endogenous cytokinins. Plant Physiol. 2003, 131, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Frebort, I.; Kowalska, M.; Hluska, T.; Frebortova, J.; Galuszka, P. Evolution of cytokinin biosynthesis and degradation. J. Exp. Bot. 2011, 62, 2431–2452. [Google Scholar] [CrossRef]
- De Lucas, M.; Davière, J.M.; Rodríguez-Falcón, M.; Pontin, M.; Iglesias-Pedraz, J.M.; Lorrain, S.; Fankhauser, C.; Blázquez, M.A.; Titarenko, E.; Prat, S. A molecular framework for light and gibberellin control of cell elongation. Nature 2008, 451, 480–484. [Google Scholar] [CrossRef]
- Que, F.; Khadr, A.; Wang, G.L.; Li, T.; Wang, Y.H.; Xu, Z.S.; Xiong, A.S. Exogenous brassinosteroids altered cell length, gibberellin content, and cellulose deposition in promoting carrot petiole elongation. Plant Sci. 2018. [Google Scholar] [CrossRef]
- Richards, D.E.; King, K.E.; Ait-ali, T.; Harberd, N.P. How gibberellin regulates plant growth and development: A molecular genetic analysis of gibberellin signaling. Annu. Rev. Plant Physiol. Plant Molec. Biol. 2001, 52, 67–88. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S. Gibberellin Metabolism and its Regulation. Annu. Rev. Plant Biol. 2008, 59, 225–251. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic. Acids. Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Overvoorde, P.J.; Okushima, Y.; Alonso, J.M.; Chan, A.; Chang, C.; Ecker, J.R.; Hughes, B.; Liu, A.; Onodera, C.; Quach, H.; et al. Functional genomic analysis of the AUXIN/INDOLE-3-ACETIC ACID gene family members in Arabidopsis thaliana. Plant Cell. 2005, 17, 3282–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, J.A.; Zenser, N.; Leyser, O.; Callis, J. Rapid degradation of auxin/indoleacetic acid proteins requires conserved amino acids of domain II and is proteasome dependent. Plant Cell. 2001, 13, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Yamamoto, K.T. Overexpression of the non-canonical Aux/IAA genes causes auxin-related aberrant phenotypes in Arabidopsis. Physiol. Plant 2008, 133, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Hagen, G.; Guilfoyle, T. Auxin-responsive gene expression: Genes, promoters and regulatory factors. Plant Mol. Biol. 2002, 49, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.B.; Hagen, G.; Guilfoyle, T. The roles of auxin response factor domains in auxin-responsive transcription. Plant Cell. 2003, 15, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, J.W. Auxin response factors. Plant Cell. Environ. 2016, 39, 1014–1028. [Google Scholar] [CrossRef] [Green Version]
- Esmon, C.A.; Tinsley, A.G.; Ljung, K.; Sandberg, G.; Hearne, L.B.; Liscum, E. A gradient of auxin and auxin-dependent transcription precedes tropic growth responses. Proc. Natl. Acad. Sci. USA 2006, 103, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, S.; Van Orden, J.; Wolf, S.; Vissenberg, K.; Delacourt, J.; Ndong, Y.A.; Pelloux, J.; Bischoff, V.; Urbain, A.; Mouille, G.; et al. A role for pectin de-methylesterification in a developmentally regulated growth acceleration in dark-grown Arabidopsis hypocotyls. New. Phytol. 2010, 188, 726–739. [Google Scholar] [CrossRef]
- Spartz, A.K.; Ren, H.; Park, M.Y.; Grandt, K.N.; Lee, S.H.; Murphy, A.S.; Sussman, M.R.; Overvoorde, P.J.; Gray, W.M. SAUR Inhibition of PP2C-D Phosphatases Activates Plasma Membrane H+-ATPases to Promote Cell Expansion in Arabidopsis. Plant Cell. 2014, 26, 2129–2142. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, D.J. Plant expansins: Diversity and interactions with plant cell walls. Curr. Opin. Plant Biol. 2015, 25, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Torii, Y.; Morita, S.; Masumura, T.; Satoh, S. Differential expression of genes identified by suppression subtractive hybridization in petals of opening carnation flowers. J. Exp. Bot. 2010, 61, 2345–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyodo, H.; Yamakawa, S.; Takeda, Y.; Tsuduki, M.; Yokota, A.; Nishitani, K.; Kohchi, T. Active gene expression of a xyloglucan endotransglucosylase/hydrolase gene, XTH9, in inflorescence apices is related to cell elongation in Arabidopsis thaliana. Plant Mol. Biol. 2003, 52, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Catalysts of plant cell wall loosening. F1000Res 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Xu, Y.; Yu, C.; He, K.; Tang, Q.; Jia, C.; He, G.; Wang, X.; Kong, Y.; Zhou, G. Transcriptome analysis of genes involved in secondary cell wall biosynthesis in developing internodes of Miscanthus lutarioriparius. Sci. Rep. 2017, 7, 9034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef]
- Wolf, S.; Hématy, K.; Höfte, H. Growth control and cell wall signaling in plants. Annu. Rev. Plant Biol. 2012, 63, 381–407. [Google Scholar] [CrossRef] [Green Version]
- Marzec-Schmidt, K.; Ludwików, A.; Wojciechowska, N.; Kasprowicz-Maluśki, A.; Mucha, J.; Bagniewska-Zadworna, A. Xylem Cell Wall Formation in Pioneer Roots and Stems of Populus trichocarpa (Torr. & Gray). Front. Plant Sci. 2019, 10, 1419. [Google Scholar] [CrossRef]
- Somerville, C. Cellulose synthesis in higher plants. Annu. Rev. Cell. Dev. Biol. 2006, 22, 53–78. [Google Scholar] [CrossRef]
- Cosgrove, D.J.; Li, L.C.; Cho, H.T.; Hoffmann-Benning, S.; Moore, R.C.; Blecker, D. The growing world of expansins. Plant Cell. Physiol. 2002, 43, 1436–1444. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.C.; Terneus, K.; Hall, Q.; Tan, L.; Wang, Y.; Wegenhart, B.L.; Chen, L.; Lamport, D.T.; Chen, Y.; Kieliszewski, M.J. Self-assembly of the plant cell wall requires an extensin scaffold. Proc. Natl. Acad. Sci. USA 2008, 105, 2226–2231. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jones, L.; McQueen-Mason, S. Expansins and cell growth. Curr. Opin. Plant Biol. 2003, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- McQueenmason, S.; Durachko, D.M.; Cosgrove, D.J. 2 Endogenous Proteins That Induce Cell-Wall Extension in Plants. Plant Cell. 1992, 4, 1425–1433. [Google Scholar] [CrossRef] [Green Version]
- McQueen-Mason, S.J.; Cosgrove, D.J. Expansin mode of action on cell walls. Analysis of wall hydrolysis, stress relaxation, and binding. Plant Physiol. 1995, 107, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQueen-Mason, S.J.; Fry, S.C.; Durachko, D.M.; Cosgrove, D.J. The relationship between xyloglucan endotransglycosylase and in-vitro cell wall extension in cucumber hypocotyls. Planta 1993, 190, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Braun, N.; Wyrzykowska, J.; Muller, P.; David, K.; Couch, D.; Perrot-Rechenmann, C.; Fleming, A.J. Conditional Repression of AUXIN BINDING PROTEIN1 Reveals That It Coordinates Cell Division and Cell Expansion during Postembryonic Shoot Development in Arabidopsis and Tobacco. Plant Cell. 2008, 20, 2746–2762. [Google Scholar] [CrossRef] [Green Version]
- Barbez, E.; Dünser, K.; Gaidora, A.; Lendl, T.; Busch, W. Auxin steers root cell expansion via apoplastic pH regulation in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2017, 114, E4884–E4893. [Google Scholar] [CrossRef] [Green Version]
- Paque, S.; Mouille, G.; Grandont, L.; Alabadí, D.; Gaertner, C.; Goyallon, A.; Muller, P.; Primard-Brisset, C.; Sormani, R.; Blázquez, M.A.; et al. AUXIN BINDING PROTEIN1 links cell wall remodeling, auxin signaling, and cell expansion in arabidopsis. Plant Cell. 2014, 26, 280–295. [Google Scholar] [CrossRef] [Green Version]
- Schopfer, P.; Liszkay, A.; Bechtold, M.; Frahry, G.; Wagner, A. Evidence that hydroxyl radicals mediate auxin-induced extension growth. Planta 2002, 214, 821–828. [Google Scholar] [CrossRef]
- Spartz, A.K.; Lee, S.H.; Wenger, J.P.; Gonzalez, N.; Itoh, H.; Inze, D.; Peer, W.A.; Murphy, A.S.; Overvoorde, P.J.; Gray, W.M. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012, 70, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Vanhaeren, H.; Gonzalez, N.; Coppens, F.; De Milde, L.; Van Daele, T.; Vermeersch, M.; Eloy, N.B.; Storme, V.; Inzé, D. Combining growth-promoting genes leads to positive epistasis in Arabidopsis thaliana. eLife 2014, 3, e02252. [Google Scholar] [CrossRef]
- Vi, S.L.; Trost, G.; Lange, P.; Czesnick, H.; Rao, N.; Lieber, D.; Laux, T.; Gray, W.M.; Manley, J.L.; Groth, D.; et al. Target specificity among canonical nuclear poly(A) polymerases in plants modulates organ growth and pathogen response. Proc. Natl. Acad. Sci. USA 2013, 110, 13994–13999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, K.; Isaacs, C.G.; Reeves, P.H.; Maloney, G.S.; Muday, G.K.; Nagpal, P.; Reed, J.W. Arabidopsis SMALL AUXIN UP RNA63 promotes hypocotyl and stamen filament elongation. Plant J. 2012, 71, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Franklin, K.A.; Lee, S.H.; Patel, D.; Kumar, S.V.; Spartz, A.K.; Gu, C.; Ye, S.; Yu, P.; Breen, G.; Cohen, J.D.; et al. PHYTOCHROME-INTERACTING FACTOR 4 (PIF4) regulates auxin biosynthesis at high temperature. Proc. Natl. Acad. Sci. USA 2011, 108, 20231–20235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, H.; Gray, W.M. SAUR Proteins as Effectors of Hormonal and Environmental Signals in Plant Growth. Mol. Plant 2015, 8, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Stortenbeker, N.; Bemer, M. The SAUR gene family: The plant’s toolbox for adaptation of growth and development. J. Exp. Bot. 2019, 70, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Arsuffi, G.; Braybrook, S.A. Acid growth: An ongoing trip. J. Exp. Bot. 2018, 69, 137–146. [Google Scholar] [CrossRef]
- Majda, M.; Robert, S. The Role of Auxin in Cell Wall Expansion. Int. J. Mol. Sci. 2018, 951. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Hayashi, K.-i.; Kinoshita, T. Auxin Activates the Plasma Membrane H+-ATPase by Phosphorylation during Hypocotyl Elongation in Arabidopsis. Plant Physiol. 2012, 159, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.I.; Takahashi, K.; Okumura-Noda, H.; Kinoshita, T. Auxin Influx Carrier AUX1 Confers Acid Resistance for Arabidopsis Root Elongation Through the Regulation of Plasma Membrane H+-ATPase. Plant Cell. Physiol. 2016, 57, 2194–2201. [Google Scholar] [CrossRef] [Green Version]
- Farquharson, K.L. SAUR19 Links Auxin and Plasma Membrane H+-ATPases in Cell Expansion. Plant Cell. 2014, 26, 1835. [Google Scholar] [CrossRef] [Green Version]
- Coenen, C.; Bierfreund, N.; Lüthen, H.; Neuhaus, G. Developmental regulation of H+-ATPase-dependent auxin responses in the diageotropica mutant of tomato (Lycopersicon esculentum). Physiol. Plant 2002, 114, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. Recent research progress on acid-growth theory. J. Exp. Bot. 2016, 43, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Nishitani, K.; Vissenberg, K. Roles of the XTH Protein Family in the Expanding Cell. Plant Cell. Monogr. 2006. [Google Scholar] [CrossRef] [Green Version]
- Hocq, L.; Pelloux, J.; Lefebvre, V. Connecting Homogalacturonan-Type Pectin Remodeling to Acid Growth. Trends Plant Sci. 2017, 22, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Knöller, A.S.; Blakeslee, J.J.; Richards, E.L.; Peer, W.A.; Murphy, A.S. Brachytic2/ZmABCB1 functions in IAA export from intercalary meristems. J. Exp. Bot. 2010, 61, 3689–3696. [Google Scholar] [CrossRef] [Green Version]
- Galuszka, P.; Frebortova, J.; Werner, T.; Yamada, M.; Strnad, M.; Schmulling, T.; Frebort, I. Cytokinin oxidase/dehydrogenase genes in barley and wheat - Cloning and heterologous expression. Eur. J. Biochem. 2004, 271, 3990–4002. [Google Scholar] [CrossRef]
- Mrizova, K.; Jiskrova, E.; Vyroubalova, S.; Novak, O.; Ohnoutkova, L.; Pospisilova, H.; Frebort, I.; Harwood, W.A.; Galuszka, P. Overexpression of Cytokinin Dehydrogenase Genes in Barley (Hordeum vulgare cv. Golden Promise) Fundamentally Affects Morphology and Fertility. PLoS ONE 2013, 8, e79029. [Google Scholar] [CrossRef] [Green Version]
- Werner, T.; Motyka, V.; Strnad, M.; Schmülling, T. Regulation of plant growth by cytokinin. Proc. Natl. Acad. Sci. USA 2001, 98, 10487–10492. [Google Scholar] [CrossRef] [Green Version]
- Koellmer, I.; Novak, O.; Strnad, M.; Schmuelling, T.; Werner, T. Overexpression of the cytosolic cytokinin oxidase/dehydrogenase (CKX7) from Arabidopsis causes specific changes in root growth and xylem differentiation. Plant J. 2014, 78, 359–371. [Google Scholar] [CrossRef]
- Efroni, I.; Blum, E.; Goldshmidt, A.; Eshed, Y. A Protracted and Dynamic Maturation Schedule Underlies Arabidopsis Leaf Development. Plant Cell. 2008, 20, 2293–2306. [Google Scholar] [CrossRef] [Green Version]
- Skalak, J.; Vercruyssen, L.; Claeys, H.; Hradilova, J.; Cerny, M.; Novak, O.; Plackova, L.; Saiz-Fernandez, I.; Skalakova, P.; Coppens, F.; et al. Multifaceted activity of cytokinin in leaf development shapes its size and structure in Arabidopsis. Plant J. 2019, 97, 805–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, A.; Clabaugh, I.; To, J.P.; Maxwell, B.B.; Chiang, Y.-H.; Schaller, G.E.; Loraine, A.; Kieber, J.J. Identification of Cytokinin-Responsive Genes Using Microarray Meta-Analysis and RNA-Seq in Arabidopsis. Plant Physiol. 2013, 162, 272–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lin, S.; Liu, D.; Gan, L.; McAvoy, R.; Ding, J.; Li, Y. Evolution and roles of cytokinin genes in angiosperms 1: Do ancient IPTs play housekeeping while non-ancient IPTs play regulatory roles? Hortic. Res. 2020, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Gazzarrini, S.; McCourt, P. Cross-talk in plant hormone signalling: What Arabidopsis mutants are telling us. Ann. Bot. 2003, 91, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, C.M.; Fishman, L. Cytoplasmic male sterility in Mimulus hybrids has pleiotropic effects on corolla and pistil traits. Heredity 2011, 106, 886–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, D.; Halevy, A.H. Stamens and gibberellin in the regulation of corolla pigmentation and growth in Petunia hybrida. Planta 1989, 179, 89–96. [Google Scholar] [CrossRef]
- Cheng, H.; Qin, L.J.; Lee, S.C.; Fu, X.D.; Richards, D.E.; Cao, D.N.; Luo, D.; Harberd, N.P.; Peng, J.R. Gibberellin regulates Arabidopsis floral development via suppression of DELLA protein function. Development 2004, 131, 1055–1064. [Google Scholar] [CrossRef] [Green Version]
- Greenboim-Wainberg, Y.; Maymon, I.; Borochov, R.; Alvarez, J.; Olszewski, N.; Ori, N.; Eshed, Y.; Weiss, D. Cross talk between gibberellin and cytokinin: The Arabidopsis GA response inhibitor SPINDLY plays a positive role in cytokinin signaling. Plant Cell. 2005, 17, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Jasinski, S.; Piazza, P.; Craft, J.; Hay, A.; Woolley, L.; Rieu, I.; Phillips, A.; Hedden, P.; Tsiantis, M. KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 2005, 15, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Yanai, O.; Shani, E.; Dolezal, K.; Tarkowski, P.; Sablowski, R.; Sandberg, G.; Samach, A.; Ori, N. Arabidopsis KNOXI proteins activate cytokinin biosynthesis. Curr. Biol. 2005, 15, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic. Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, B.; Du, M.; Eneji, A.E.; Wang, B.; Duan, L.; Li, Z.; Tian, X. Mechanism of phytohormone involvement in feedback regulation of cotton leaf senescence induced by potassium deficiency. J. Exp. Bot. 2012, 63, 5887–5901. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | logFC | p value | Annotation |
|---|---|---|---|
| Cell expansion (stage 3) | |||
| Solyc02g080220.2 | 1.61 | 4.34 × 10−53 | Pectinesterase |
| Solyc07g052980.2 | −1.30 | 3.22 × 10−33 | Xyloglucan endotransglucosylase/hydrolase protein 9 |
| Solyc08g005800.2 | −1.71 | 2.64 × 10−36 | Pectin acetylesterase |
| Solyc07g043390.2 | −1.79 | 1.11 × 10−90 | cellulose synthase-like protein G2 |
| Solyc02g088100.2 | −1.99 | 4.22 × 10−47 | Expansin-A5 |
| Solyc02g078040.2 | −2.16 | 6.28 × 10−60 | pistil-specific extensin-like protein |
| Solyc05g007830.2 | −2.39 | 1.79× 10−59 | Expansin-A15 |
| Solyc05g014000.2 | −2.78 | 2.60× 10−180 | Pectate lyase |
| Solyc03g083770.1 | −4.14 | 0 | pectin methylesterase inhibitor |
| Solyc06g005560.2 | −4.60 | 1.83 × 10−35 | Expansin9 |
| Cell expansion (stage 4) | |||
| Solyc01g106650.2 | 3.28 | 9.68 × 10−58 | Xyloglucan endotransglucosylase/hydrolase protein 10 |
| Solyc06g034370.1 | 1.64 | 6.36 × 10−134 | pectin methylesterase inhibitor |
| Solyc03g097050.2 | 1.46 | 4.00 × 10−65 | cellulose synthase-like protein D3 |
| Solyc07g017600.2 | 1.28 | 7.32 × 10−60 | Pectinesterase |
| Solyc04g074290.2 | 1.28 | 2.21 × 10−74 | pectin methyltransferase QUA2 |
| Solyc03g083730.1 | −1.08 | 1.76 × 10−59 | pectin methylesterase inhibitor |
| Solyc03g025600.2 | −1.17 | 4.64 × 10−62 | Pectin acetylesterase |
| Solyc09g097770.2 | −1.34 | 1.22 × 10−168 | Cell wall protein |
| Solyc08g077330.2 | −2.44 | 2.38× 10−107 | expansin-like B1 |
| Solyc06g084620.1 | −2.86 | 2.12 × 10−48 | Pectinesterase |
| Auxin (stage 3) | |||
| Solyc05g025920.2 | 8.66 | 1.98 × 10−8 | Auxin-induced protein 15A |
| Solyc06g075690.2 | 2.23 | 1.51 × 10−91 | Auxin-regulated protein |
| Solyc09g075210.2 | 1.08 | 3.11 × 10−32 | indole-3-acetic acid-induced protein ARG2 |
| Solyc02g077880.2 | 1.05 | 1.14 × 10−36 | Auxin repressed/dormancy associated protein |
| Solyc10g008520.2 | −1.36 | 1.43 × 10−29 | Indole-3-acetic acid-amido synthetase GH3.10 |
| Solyc11g013310.1 | −1.91 | 2.97 × 10−37 | Auxin transporter-like protein 3 |
| Solyc03g123410.1 | −1.96 | 6.59 × 10−35 | auxin-binding protein ABP19a |
| Solyc11g069190.1 | −2.00 | 2.29 × 10−35 | Auxin response factor |
| Solyc09g056390.1 | −2.13 | 2.09 × 10−66 | auxin-induced in root cultures protein 12 |
| Solyc07g041720.1 | −2.99 | 2.45 × 10−162 | auxin-binding protein ABP19a-like |
| Auxin (stage 4) | |||
| Solyc07g066560.1 | 2.29 | 5.62 × 10−30 | auxin-responsive protein SAUR71 |
| Solyc06g075690.2 | 2.14 | 0 | Auxin-regulated protein |
| Solyc12g014500.1 | 1.87 | 2.91 × 10−13 | indole-3-acetate O-methyltransferase 1-like |
| Solyc04g081240.2 | 1.07 | 1.16 × 10−6 | Auxin response factor 5 |
| Solyc04g076850.2 | −1.14 | 1.62 × 10−94 | Auxin-responsive protein |
| Solyc09g075210.2 | −1.35 | 2.30 × 10−182 | indole-3-acetic acid-induced protein ARG2 |
| Solyc02g079190.2 | −1.64 | 5.46 × 10−122 | protein AUXIN SIGNALING F-BOX 2 |
| Solyc06g063060.2 | −2.06 | 3.94 × 10−21 | Auxin repressed protein |
| Solyc09g083290.2 | −2.79 | 1.06 × 10−35 | Auxin-responsive protein |
| Solyc06g053840.2 | −2.79 | 3.09 × 10−34 | Auxin-responsive protein |
| Gibberellins (stage 3) | |||
| Solyc02g070430.2 | 6.31 | 2.08 × 10−24 | gibberellin 2-beta-dioxygenase 1 |
| Solyc12g042500.1 | 2.63 | 3.77 × 10−08 | Gibberellin regulated protein |
| Solyc07g061730.2 | 2.32 | 1.09 × 10−20 | Gibberellin 2-oxidase |
| Solyc07g056670.2 | 1.93 | 1.71 × 10−15 | Gibberellin 2-oxidase 2 |
| Solyc01g111080.2 | 1.59 | 2.12 × 10−43 | Gibberellin-regulated protein 1 |
| Solyc03g113910.2 | 1.42 | 2.34 × 10−06 | gibberellin-regulated protein 10 |
| Solyc06g008870.2 | 1.11 | 4.70 × 10−12 | gibberellin receptor GID1B-like |
| Solyc02g089350.2 | −2.50 | 8.37 × 10−46 | Gibberellin regulated protein |
| Solyc03g119910.2 | −3.83 | 7.14 × 10−15 | Gibberellin 3-beta-dioxygenase 1 |
| Solyc03g116060.2 | −5.80 | 5.10 × 10−4 | Gibberellin-regulated protein 4 |
| Gibberellins (stage 4) | |||
| Solyc12g042500.1 | 2.48 | 2.00 × 10−4 | Gibberellin regulated protein |
| Solyc04g078200.2 | 1.95 | 2.94 × 10−27 | gibberellin-regulated family protein precursor |
| Solyc03g116060.2 | 1.70 | 7.30 × 10−3 | Gibberellin-regulated protein 4 |
| Solyc06g007890.2 | 1.52 | 1.56 × 10−2 | Gibberellin regulated protein |
| Solyc07g063940.1 | 1.28 | 5.31 × 10−62 | Chitin-inducible gibberellin-responsive protein 1 |
| Solyc01g111080.2 | 1.15 | 2.47 × 10−198 | Gibberellin-regulated protein 1 |
| Solyc07g061720.2 | −1.02 | 8.70 × 10−4 | Gibberellin 2-oxidase |
| Solyc11g072310.1 | −1.54 | 1.00 × 10−2 | Gibberellin 20-oxidase-3 |
| Solyc07g061730.2 | −3.96 | 7.97 × 10−18 | Gibberellin 2-oxidase |
| Cytokinin (stage3) | |||
| Solyc04g014990.1 | 3.46 | 1.82 × 10−8 | zeatin O-xylosyltransferase-like |
| Solyc01g088160.2 | 2.71 | 3.54 × 10−94 | Cytokinin oxidase/dehydrogenase-like |
| Solyc10g079930.1 | 2.25 | 2.63 × 10−48 | zeatin O-glucosyltransferase-like |
| Solyc06g062330.1 | 2.05 | 5.77 × 10−19 | zeatin O-glucosyltransferase-like |
| Solyc04g081290.2 | 2.00 | 9.30 × 10−19 | Cytokinin riboside 5′-monophosphate phosphoribohydrolase |
| Solyc04g008310.1 | 1.67 | 5.45 × 10−38 | zeatin O-xylosyltransferase-like |
| Solyc08g062820.2 | 1.56 | 7.56 × 10−31 | Cytokinin riboside 5′-monophosphate phosphoribohydrolase |
| Solyc12g008900.1 | 1.74 | 2.16 × 10−8 | cytokinin dehydrogenase 3 isoform X1 |
| Solyc05g053400.1 | −1.95 | 1.05 × 10−18 | zeatin O-xylosyltransferase-like |
| Solyc11g066670.1 | −2.16 | 1.66 × 10−56 | zeatin O-glucosyltransferase-like |
| Cytokinin (stage4) | |||
| Solyc01g088160.2 | 1.79 | 5.20 × 10−18 | Cytokinin oxidase/dehydrogenase-like |
| Solyc06g075090.2 | 1.61 | 1.25 × 10−6 | Cytokinin riboside 5′-monophosphate phosphoribohydrolase |
| Solyc06g062330.1 | −1.18 | 1.60 × 10−3 | zeatin O-glucosyltransferase-like |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Shi, M.; Wang, J.; Zhang, B.; Li, Y.; Wang, J.; El-Sappah, A.H.; Liang, Y. Comparative Transcriptomic Analysis of the Development of Sepal Morphology in Tomato (Solanum Lycopersicum L.). Int. J. Mol. Sci. 2020, 21, 5914. https://doi.org/10.3390/ijms21165914
Liu J, Shi M, Wang J, Zhang B, Li Y, Wang J, El-Sappah AH, Liang Y. Comparative Transcriptomic Analysis of the Development of Sepal Morphology in Tomato (Solanum Lycopersicum L.). International Journal of Molecular Sciences. 2020; 21(16):5914. https://doi.org/10.3390/ijms21165914
Chicago/Turabian StyleLiu, Jingyi, Meijing Shi, Jing Wang, Bo Zhang, Yushun Li, Jin Wang, Ahmed. H. El-Sappah, and Yan Liang. 2020. "Comparative Transcriptomic Analysis of the Development of Sepal Morphology in Tomato (Solanum Lycopersicum L.)" International Journal of Molecular Sciences 21, no. 16: 5914. https://doi.org/10.3390/ijms21165914
APA StyleLiu, J., Shi, M., Wang, J., Zhang, B., Li, Y., Wang, J., El-Sappah, A. H., & Liang, Y. (2020). Comparative Transcriptomic Analysis of the Development of Sepal Morphology in Tomato (Solanum Lycopersicum L.). International Journal of Molecular Sciences, 21(16), 5914. https://doi.org/10.3390/ijms21165914