Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies

 , , ,
, , ,
Abstract
:1. Introduction
2. IL-1 Receptor Family and Its Ligands
3. IL-1: Active Precursors and Dual Function
4. Role of IL-1 in Solid Tumors
4.1. IL-1 Is an Inducer of Carcinogenesis
4.2. IL-1 Mediates Angiogenesis and Metastasis
4.3. IL-1 Is Responsible for Immunosuppression
5. IL-1 and Resistance to Targeted Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IL-1 | Interleukin-1 |
| TAM | Tumor-associated macrophage |
| MDSC | Myeloid-derived suppressor cell |
| IL-1R | Interleukin-1 receptor |
| Th17 | T helper 17 |
| Treg | Regulatory T cell |
| Breg | Regulatory B cell |
| TLR | Toll like receptor |
| MyD88 | Myeloid differentiation primary response 88 |
| VEGF | Vascular endothelial growth factor |
| TAN | Tumor-associated neutrophil |
| EC | Endothelial cell |
References
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Voronov, E. Immunotherapeutic approaches of IL-1 neutralization in the tumor microenvironment. J. Leukoc. Biol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Nicolini, A.; Ferrari, P.; Rossi, G.; Carpi, A. Tumour growth and immune evasion as targets for a new strategy in advanced cancer. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018. [Google Scholar] [CrossRef]
- Voronov, E.; Dotan, S.; Krelin, Y.; Song, X.; Elkabets, M.; Carmi, Y.; Rider, P.; Cohen, I.; Romzova, M.; Kaplanov, I.; et al. Unique versus redundant functions of IL-1α and IL-1β in the tumor microenvironment. Front. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.; Rider, P.; Carmi, Y.; Braiman, A.; Dotan, S.; White, M.R.; Voronov, E.; Martin, M.U.; Dinarello, C.A.; Apte, R.N.; et al. Differential release of chromatin-bound IL-1α discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [Green Version]
- Werman, A.; Werman-Venkert, R.; White, R.; Lee, J.K.; Werman, B.; Krelin, Y.; Voronov, E.; Dinarello, C.A.; Apte, R.N. The precursor form of IL-1α is an intracrine proinflammatory activator of transcription. Proc. Natl. Acad. Sci. USA 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006. [Google Scholar] [CrossRef] [PubMed]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 beta—A friend or foe in malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.; Kanneganti, T.D. Function and regulation of IL-1α in inflammatory diseases and cancer. Immunol. Rev. 2018. [Google Scholar] [CrossRef]
- Elaraj, D.M.; Weinreich, D.M.; Varghese, S.; Puhlmann, M.; Hewitt, S.M.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. 2006. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Tang, Y.; Hua, S. Immunological approaches towards cancer and inflammation: A cross talk. Front. Immunol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Fahey, E.; Doyle, S.L. IL-1 family cytokine regulation of vascular permeability and angiogenesis. Front. Immunol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and Anti-Inflammatory Equilibrium, Proliferative and Antiproliferative Balance: The Role of Cytokines in Multiple Myeloma. Mediat. Inflamm. 2017. [Google Scholar] [CrossRef] [Green Version]
- Krelin, Y.; Voronov, E.; Dotan, S.; Elkabets, M.; Reich, E.; Fogel, M.; Huszar, M.; Iwakura, Y.; Segal, S.; Dinarello, C.A.; et al. Interleukin-1β-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronov, E.; Carmi, Y.; Apte, R.N. The role IL-1 in tumor-mediated angiogenesis. Front. Physiol. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nakata, K.; Kashimoto, S.; Yoshida, H.; Yamada, M. Antitumor effect of recombinant human interleukin 1 alpha against murine syngeneic tumors. Jpn. J. Cancer Res. GANN 1986. [Google Scholar] [CrossRef]
- Haabeth, O.A.W.; Lorvik, K.B.; Yagita, H.; Bogen, B.; Corthay, A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology 2016. [Google Scholar] [CrossRef]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 family members in cancer; two sides to every story. Front. Immunol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1β promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012. [Google Scholar] [CrossRef] [Green Version]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006. [Google Scholar] [CrossRef]
- Cataisson, C.; Salcedo, R.; Hakim, S.; Moffitt, B.A.; Wright, L.; Yi, M.; Stephens, R.; Dai, R.M.; Lyakh, L.; Schenten, D.; et al. IL-1R-MyD88 signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabets, M.; Krelin, Y.; Dotan, S.; Cerwenka, A.; Porgador, A.; Lichtenstein, R.G.; White, M.R.; Zoller, M.; Iwakura, Y.; Dinarello, C.A.; et al. Host-Derived Interleukin-1α Is Important in Determining the Immunogenicity of 3-Methylcholantrene Tumor Cells. J. Immunol. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Marusawa, H.; Endo, Y.; Chiba, T. Inflammation-mediated genomic instability: Roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Apte, R.N. IL-1 in Colon Inflammation, Colon Carcinogenesis and Invasiveness of Colon Cancer. Cancer Microenviron. 2015. [Google Scholar] [CrossRef]
- Lee, J.G.; Kay, E.D.P. NF-κB is the transcription factor for FGF-2 that causes endothelial mesenchymal transformation in cornea. Investig. Ophthalmol. Vis. Sci. 2012. [Google Scholar] [CrossRef]
- Hye, W.K.; Torres, D.; Wald, L.; Weissleder, R.; Bogdanov, A.A. Targeted imaging of human endothelial-specific marker in a model of adoptive cell transfer. Lab. Investig. 2006. [Google Scholar] [CrossRef]
- Jagielska, J.; Kapopara, P.R.; Salguero, G.; Scherr, M.; Schütt, H.; Grote, K.; Schieffer, B.; Bavendiek, U. Interleukin-1β assembles a proangiogenic signaling module consisting of caveolin-1, Tumor necrosis factor receptor-associated factor 6, p38-mitogen-activated protein kinase (MAPK), and mapk-activated protein kinase 2 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Schweighofer, B.; Testori, J.; Sturtzel, C.; Sattler, S.; Mayer, H.; Wagner, O.; Bilban, M.; Hofer, E. The VEGF-induced transcriptional response comprises gene clusters at the crossroad of angiogenesis and inflammation. Thromb. Haemost. 2009. [Google Scholar] [CrossRef] [Green Version]
- Sheikpranbabu, S.; Kalishwaralal, K.; Venkataraman, D.; Eom, S.H.; Park, J.; Gurunathan, S. Silver nanoparticles inhibit VEGF-and IL-1β-induced vascular permeability via Src dependent pathway in porcine retinal endothelial cells. J. Nanobiotechnol. 2009. [Google Scholar] [CrossRef] [Green Version]
- Rider, P.; Kaplanov, I.; Romzova, M.; Bernardis, L.; Braiman, A.; Voronov, E.; Apte, R.N. The transcription of the alarmin cytokine interleukin-1 alpha is controlled by hypoxia inducible factors 1 and 2 alpha in hypoxic cells. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salven, P.; Hattori, K.; Heissig, B.; Rafii, S. Interleukin-1alpha promotes angiogenesis in vivo via VEGFR-2 pathway by inducing inflammatory cell VEGF synthesis and secretion. FASEB J. 2002. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Farnarier, C.; Kaplanski, S.; Porat, R.; Shapiro, L.; Bongrand, P.; Dinarello, C.A. Interleukin-1 induces interleukin-8 secretion from endothelial cells by a juxtacrine mechanism. Blood 1994, 84, 4242–4248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Vanaclocha, F.; Fantuzzi, G.; Mendoza, L.; Fuentes, A.M.; Anasagasti, M.J.; Martín, J.; Carrascal, T.; Walsh, P.; Reznikov, L.L.; Kim, S.H.; et al. IL-18 regulates IL-1β-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl. Acad. Sci. USA 2000. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Vanaclocha, F.; Amézaga, C.; Asumendi, A.; Kaplanski, G.; Dinarello, C.A. Interleukin-1 Receptor Blockade Reduces the Number and Size of Murine B16 Melanoma Hepatic Metastases. Cancer Res. 1994, 54, 2667–2672. [Google Scholar]
- León, X.; Bothe, C.; García, J.; Parreño, M.; Alcolea, S.; Quer, M.; Vila, L.; Camacho, M. Expression of IL-1α correlates with distant metastasis in patients with head and neck squamous cell carcinoma. Oncotarget 2015. [Google Scholar] [CrossRef] [Green Version]
- Dekker, S.K.; Vink, J.; Bruijn, J.A.; Mihm, M.C., Jr.; Vermeer, B.J.; Byers, H.R. Characterization of interleukin-1α-induced melanoma cell motility: Inhibition by type I and type II receptor-blocking monoclonal antibodies. Melanoma Res. 1997. [Google Scholar] [CrossRef]
- Filippi, I.; Carraro, F.; Naldini, A. Interleukin-1 β affects MDAMB231 breast cancer cell migration under hypoxia: Role of HIF-1 α and NF B transcription factors. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef] [Green Version]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Bradbury, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016. [Google Scholar] [CrossRef] [Green Version]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of Interleukin-1β Induces Gastric Inflammation and Cancer and Mobilizes Myeloid-Derived Suppressor Cells in Mice. Cancer Cell 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Gebhardt, C.; Umansky, L.; Beckhove, P.; Schulze, T.J.; Utikal, J.; Umansky, V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int. J. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Gebhardt, C.; Umansky, L.; Beckhove, P.; Schulze, T.J.; Utikal, J.; Umansky, V. The Role of IL-1β in the Early Tumor Cell–Induced Angiogenic Response. J. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nature Rev. Immunol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Vatan, L.; Escara-Wilke, J.; Szeliga, W.; Keller, E.T.; Zou, W. Cutting Edge: Opposite Effects of IL-1 and IL-2 on the Regulation of IL-17 + T Cell Pool IL-1 Subverts IL-2-Mediated Suppression. J. Immunol. 2007. [Google Scholar] [CrossRef] [Green Version]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.G.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006. [Google Scholar] [CrossRef]
- Michael-Robinson, J.M.; Biemer-Hüttmann, A.; Purdie, D.M.; Walsh, M.D.; Simms, L.A.; Biden, K.G.; Young, J.P.; Leggett, B.A.; Jass, J.R.; Radford-Smith, G.L. Tumour infiltrating lymphocytes and apoptosis are independent features in colorectal cancer stratified according to microsatellite instability status. Gut 2001. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Archiv. 2016. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- West, N.R.; Mccuaig, S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nature Rev. Immunol. 2015. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta-Rev. Cancer 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Garduño, A.M.; Mendoza-Rodríguez, M.G.; Urrutia-Cabrera, D.; Domínguez-Robles, M.C.; Pérez-Yépez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1β induced methylation of the estrogen receptor ERα gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, Y.N. Hypoxia/IL-1α axis promotes gastric cancer progression and drug resistance. J. Dig. Dis. 2017. [Google Scholar] [CrossRef]
- Mendoza-Rodríguez, M.G.; Ayala-Sumuano, J.T.; García-Morales, L.; Zamudio-Meza, H.; Pérez-Yepez, E.A.; Meza, I. IL-1β inflammatory cytokine-induced TP63 isoform ∆NP63α signaling cascade contributes to cisplatin resistance in human breast cancer cells. Int. J. Mol. Sci. 2019, 20, 270. [Google Scholar] [CrossRef] [Green Version]
- Maria, F.F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravatà, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamm. (United Kingdom) 2015. [Google Scholar] [CrossRef] [Green Version]
- Stanam, A.; Gibson-Corley, K.N.; Love-Homan, L.; Ihejirika, N.; Simons, A.L. Interleukin-1 blockade overcomes erlotinib resistance in head and neck squamous cell carcinoma. Oncotarget 2016. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.T.; Love-Homan, L.; Espinosa-Cotton, M.; Stanam, A.; Simons, A.L. MyD88-dependent signaling decreases the antitumor efficacy of epidermal growth factor receptor inhibition in head and neck cancer cells. Cancer Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Lan, X.; Wang, T.; Lu, H.; Cao, M.; Yan, S.; Cui, Y.; Jia, D.; Cai, L.; Xing, Y. Targeting the IL-1β/EHD1/TUBB3 axis overcomes resistance to EGFR-TKI in NSCLC. Oncogene 2020. [Google Scholar] [CrossRef]
- Gelfo, V.; Mazzeschi, M.; Grilli, G.; Lindzen, M.; Santi, S.; D’Uva, G.; Győrffy, B.; Ardizzoni, A.; Yarden, Y.; Lauriola, M. A Novel Role for the Interleukin-1 Receptor Axis in Resistance to Anti-EGFR Therapy. Cancers (Basel) 2018, 10, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfo, V.; Rodia, M.T.; Pucci, M.; Dall’Ora, M.; Santi, S.; Solmi, R.; Roth, L.; Lindzen, M.; Bonafè, M.; Bertotti, A.; et al. A module of inflammatory cytokines defines resistance of colorectal cancer to EGFR inhibitors. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.; Porciuncula, A.; Yu, A.; Iwakura, Y.; David, G. Uncoupling the Senescence-Associated Secretory Phenotype from Cell Cycle Exit via Interleukin-1 Inactivation Unveils Its Protumorigenic Role. Mol. Cell. Biol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.; Belenki, D.; Däbritz, J.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2017, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Wang, P.; Zou, Y.; Zha, Z.; Huang, H.; Guan, M.; Wu, Y.; Liu, G. IL-1β Promotes Stemness of Tumor Cells by Activating Smad/ID1 Signaling Pathway. Int. J. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Hickish, T.; Andre, T.; Wyrwicz, L.; Saunders, M.; Sarosiek, T.; Kocsis, J.; Nemecek, R.; Rogowski, W.; Lesniewski-Kmak, K.; Petruzelka, L.; et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; CANTOS Trial Group. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017. [Google Scholar] [CrossRef]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Dispenzieri, A.; Gertz, M.A.; Witzig, T.E.; Kumar, S.; Hayman, S.R.; Russell, S.J.; Buadi, F.K. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 2009. [Google Scholar] [CrossRef]
- Stewart, A.G.; Beart, P.M. Inflammation: Maladies, models, mechanisms and molecules. Br. J. Pharmacol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozinovski, S.; Vlahos, R.; Anthony, D.; McQualter, J.; Anderson, G.; Irving, L.; Steinfort, D. COPD and squamous cell lung cancer: Aberrant inflammation and immunity is the common link. Br. J. Pharmacol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. 2019. [Google Scholar] [CrossRef]
- Rogovskii, V.S. The Linkage Between Inflammation and Immune Tolerance: Interfering with Inflammation in Cancer. Curr. Cancer Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Apte, R.N. Targeting the Tumor Microenvironment by Intervention in Interleukin-1 Biology. Curr. Pharm. Des. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Borcherding, N.; Kolb, R. IL-1 Signaling in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 1–23. [Google Scholar]
- Cui, G.; Yuan, A.; Sun, Z.; Zheng, W.; Pang, Z. IL-1β/IL-6 network in the tumor microenvironment of human colorectal cancer. Pathol. Res. Pract. 2018. [Google Scholar] [CrossRef]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ Immature Myeloid Cells Mediate Suppression of T Cells in Mice Bearing Tumors of IL-1β-Secreting Cells. J. Immunol. 2005. [Google Scholar] [CrossRef] [Green Version]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation Induces Myeloid-Derived Suppressor Cells that Facilitate Tumor Progression. J. Immunol. 2006. [Google Scholar] [CrossRef] [Green Version]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory Cytokine IL-1β Promotes Tumor Growth of Lewis Lung Carcinoma by Induction of Angiogenic Factors: In Vivo Analysis of Tumor-Stromal Interaction. J. Immunol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013. [Google Scholar] [CrossRef] [PubMed]


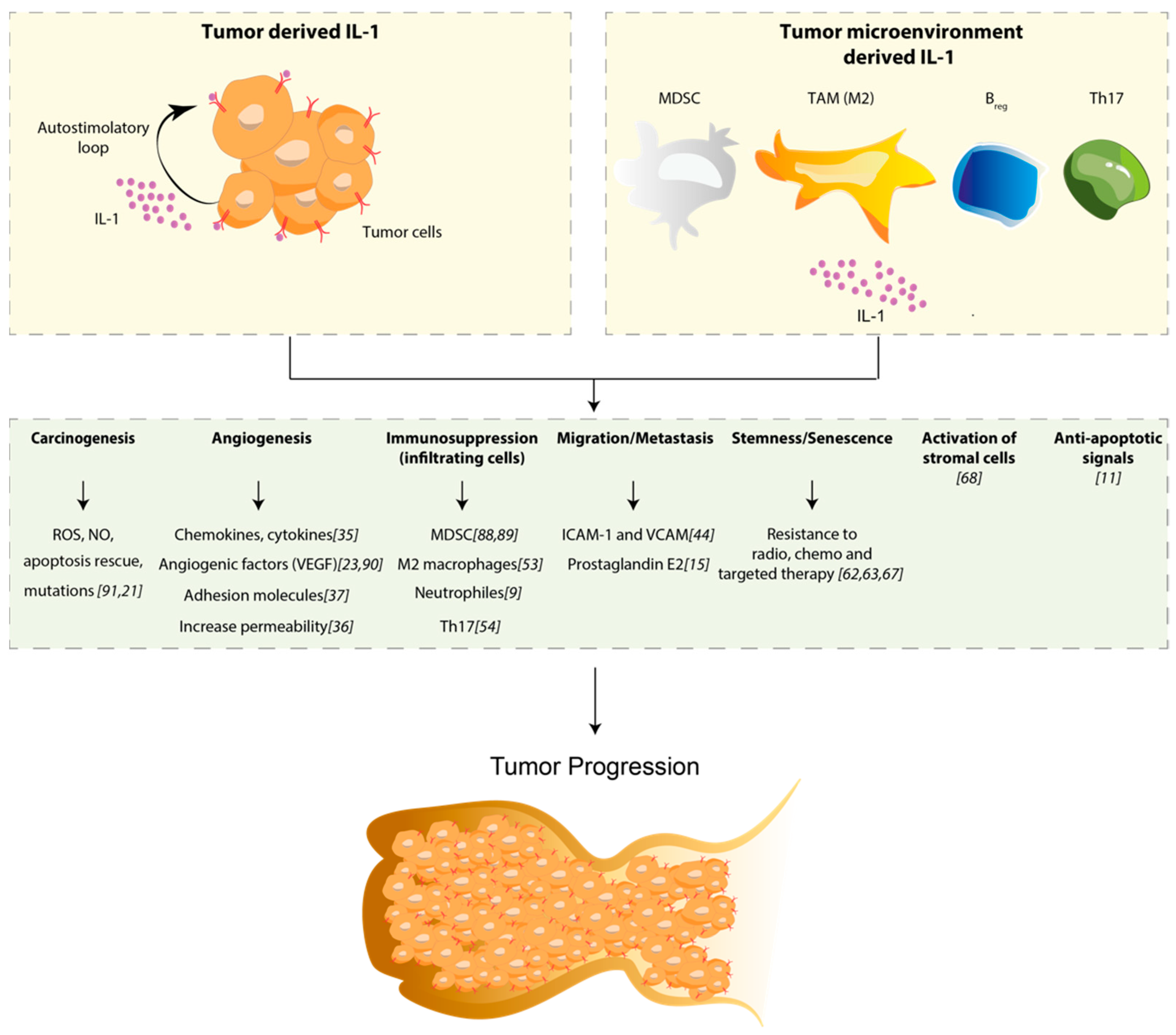{kind=link}
| Therapy | Target | Tumor Type | Recruitment Status | Development status | ClinicalTrial.gov Identifier | Sponsor | Start Date | Estimated Completition Date |
|---|---|---|---|---|---|---|---|---|
| Anakinra + Everolimus | IL-1 Ra + anti mTOR | Neoplasm | Active, not recruiting | Phase 1 | NCT01624766 | M.D. Anderson Cancer Center | June, 2012 | June, 2020 |
| Anakinra + Chemo | IL-1 Ra + anti mTOR | Pancreatic Adenocarcinoma | Active, not recruiting | Early Phase 1 | NCT02550327 | Baylor Research Institute | January, 2016 | August, 2023 |
| Anakinra + JCARH125 | IL-1 Ra + CAR T-cells | Multiple Myeloma | Recruiting | Phase 2 | NCT03430011 | Juno Therapeutics | March, 2023 | March, 2023 |
| Anakinra | IL-1 Ra | Multiple Myeloma | Active, not recruiting | Phase 2 | NCT03233776 | Radboud University | May, 2019 | June, 2020 |
| Anakinra | IL-1 Ra | Multiple Myeloma | Recruiting | Phase 2 | NCT04099901 | Radboud University | October, 2020 | October, 2022 |
| Anakinra + Axicabtagene Ciloleucel | IL-1 Ra + CAR T-cells | Neoplasm, Large B-Cell Lymphoma | Not yet recruiting | Phase 1,2 | NCT04432506 | M.D. Anderson Cancer Center | July, 2020 | January, 2025 |
| Anakinra + Axicabtagene Ciloleucel | IL-1 Ra + CAR T-cells | B-Cell Non-Hodgkin Lymphoma | Not yet recruiting | Phase 2 | NCT04359784 | Fred Hutchinson Cancer Research Center | August, 2020 | December, 2021 |
| Anakinra + Axicabtagene Ciloleucel | IL-1 Ra + CAR T-cells | Non-Hodgkin Lymphoma | Not yet recruiting | Phase 2 | NCT04150913 | Marcela V. Maus, M.D.;Ph.D. | July, 2020 | November, 2024 |
| Anakinra | IL-1 Ra | B-Cell Lymphoma and Non-Hodgkin Lymphoma | Recruiting | Phase 2 | NCT04148430 | Memorial Sloan Kettering Cancer Center | October, 2019 | October, 2022 |
| Anakinra + Axicabtagene Ciloleucel | IL-1 Ra + CAR T-cells | Large B-Cell Lymphoma | Recruiting | Phase 2 | NCT04205838 | Jonsson Comprehensive Cancer Center | March, 2020 | December, 2022 |
| Canakinumab | mAb anti IL-1β | Non-small Cell Lung Cancer | Recruiting | Phase 3 | NCT03447769 | Novartis Pharmaceuticals | March, 2018 | January, 2027 |
| Canakinumab + Spartalizumab + LAG525 | mAb anti IL-1β + mAb anti PD-1+ mAb anti LAG-3 | Triple Negative Breast Cancer | Recruiting | Phase 1 | NCT03742349 | Novartis Pharmaceuticals | January, 2019 | January, 2022 |
| Anakinra +/− Pembrolizumab | mAb anti IL-1β +/− mAb anti PDL-1 | Non-small Cell Lung Cancer | Recruiting | Phase 2 | NCT03968419 | Novartis Pharmaceuticals | November, 2019 | January, 2022 |
| Anakinra + Pembrolizumab + Chemo | mAb anti IL-1β +/− mAb anti PDL-1 | Non-small Cell Lung Cancer | Active, not recruiting | Phase 3 | NCT03631199 | Novartis Pharmaceuticals | December, 2018 | September, 2022 |
| Canakinumab + PDR001 | mAb anti IL-1β + mAb anti PD-1 | Triple Negative Breast Cancer and NSCLC | Active, not recruiting | Phase 1 | NCT02900664 | Novartis Pharmaceuticals | August, 2016 | August, 2020 |
| Canakinumab + Spartalizumab | mAb anti IL-1β + mAb anti PD-1 | Renal Cell Carcinoma | Recruiting | Early Phase 1 | NCT04028245 | Charles G. Drake | August, 2019 | December, 2021 |
| Canakinumab | mAb anti IL-1β | Myelodysplastic Syndrome or Chronic Myelomonocytic Leukemia | Not yet recruiting | Phase 2 | NCT04239157 | M.D. Anderson Cancer Center | June, 2020 | December, 2021 |
| Canakinumab + PDR001 + Chemo | mAb anti IL-1β + mAb anti PD-1 | Non-small Cell Lung Cancer | Active, not recruiting | Phase 1 | NCT03064854 | Novartis Pharmaceuticals | May, 2017 | December, 2021 |
| Canakinumab + Spartalizumab | mAb anti IL-1β + mAb anti PD-1 | Melanoma | Recruiting | Phase 2 | NCT03484923 | Novartis Pharmaceuticals | September, 2018 | June, 2022 |
| Canakinumab + Chemo | mAb anti IL-1β | Non-small Cell Lung Cancer | Active, not recruiting | Phase 3 | NCT03626545 | Novartis Pharmaceuticals | January, 2019 | March, 2022 |
| Xilonix + Chemo | mAb anti IL-1α | Pancreatic cancer | Active, not recruiting | Phase 1 | NCT03207724 | Andrew Hendifar, MD | October, 2017 | December, 2020 |
| CAN04 + Pembrolizumab | mAb anti IL1RAP+mAb anti PD-1 | Non-Small-Cell Lung, Urothelial CarcinomaMalignant Melanoma, Head and Neck Squamous Cell Carcinoma | Not yet recruiting | Phase 1 | NCT04452214 | Cantargia AB | September, 2020 | January, 2022 |
| CAN04 + Chemo | mAb anti IL1RAP | Non Small Cell Lung Cancer, Pancreatic Ductal Adenocarcinoma, Triple Negative Breast Cancer, Colorectal Cancer | Recruiting | Phase 1/2 | NCT03267316 | Cantargia AB | September, 2017 | June, 2021 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. https://doi.org/10.3390/ijms21176009
Gelfo V, Romaniello D, Mazzeschi M, Sgarzi M, Grilli G, Morselli A, Manzan B, Rihawi K, Lauriola M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. International Journal of Molecular Sciences. 2020; 21(17):6009. https://doi.org/10.3390/ijms21176009
Chicago/Turabian StyleGelfo, Valerio, Donatella Romaniello, Martina Mazzeschi, Michela Sgarzi, Giada Grilli, Alessandra Morselli, Beatrice Manzan, Karim Rihawi, and Mattia Lauriola. 2020. "Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies" International Journal of Molecular Sciences 21, no. 17: 6009. https://doi.org/10.3390/ijms21176009
APA StyleGelfo, V., Romaniello, D., Mazzeschi, M., Sgarzi, M., Grilli, G., Morselli, A., Manzan, B., Rihawi, K., & Lauriola, M. (2020). Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. International Journal of Molecular Sciences, 21(17), 6009. https://doi.org/10.3390/ijms21176009