Extracellular Vesicles in the Development of Cancer Therapeutics



Abstract
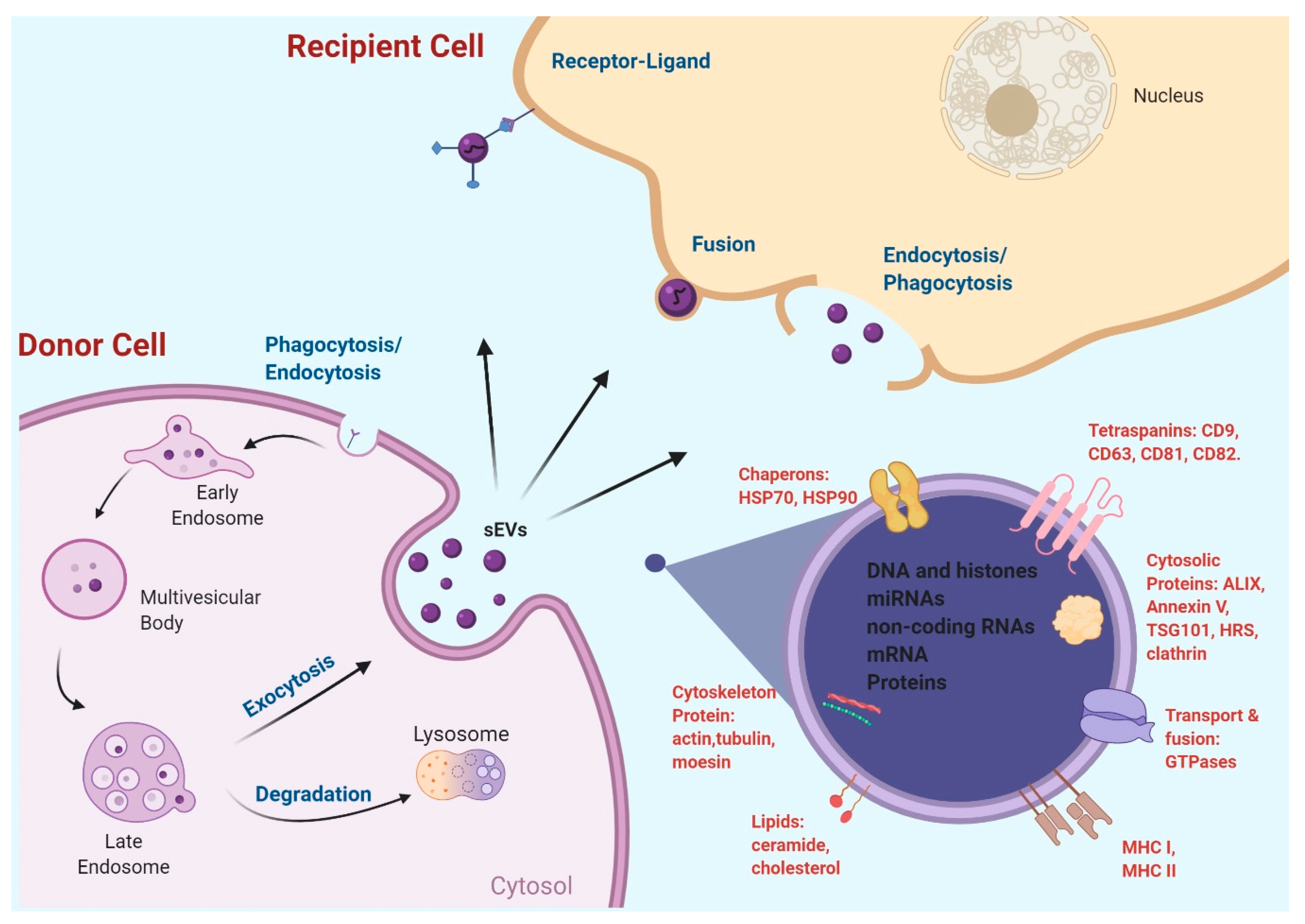
:1. Introduction
2. EV Cargos and Functions
2.1. EV Nomenclature
2.2. EV Surface Markers and Cargos
2.3. EV Functions
3. EVs as Potential Therapeutic Targets in Cancer
3.1. Suppressing sEV Biogenesis and Release
3.2. Preventing EV Uptake
3.3. Eliminating Circulating Cancer sEVs
3.4. Targeting Specific sEV Cargo Components
4. EVs as Drug Carriers in Cancer Treatment
4.1. EV Sources and Loading Efficiency
4.2. Loading Therapeutics into sEVs via Donor Cells
4.3. Loading Therapeutics into Isolated sEVs
5. Clinical Trials Testing sEVs as Cancer Therapeutic Carriers
5.1. Clinical Trials Using Dendritic Cell-Derived sEVs (DEX)
5.2. Clinical Trials Using Ascites-Derived sEVs (AEX)
5.3. Clinical Trials Using Tumor Cell-Derived EVs
5.4. Clinical Trials Using Plant-Derived sEVs
5.5. Clinical Trials Using Normal Fibroblast-Like Mesenchymal Cell-Derived EVs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EV | Extracellular Vesicle |
| MV | Microvesicle |
| ISEV | International Society for Extracellular Vesicles |
| sEV | Small Extracellular Vesicle |
| MHC | Major Histocompatibility Complex |
| EPCAM | Epithelial Cell Adhesion Molecule |
| PECAM1 | Platelet Endothelial Cell Adhesion Molecule 1 |
| ERBB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| ESCRT | Endosomal Sorting Complexes Required for Transport |
| nSMase2 | Neutral sphingomyelinase 2 |
| HSP72 | Heat-Shock Protein 72 |
| STAT-3 | Signal Transducer and Activator of Transcription 3 |
| MA | Manumycin-A |
| PI3K | Phosphatidylinositol 3-Kinase |
| HUVEC | Human Umbilical Vein Endothelial Cell |
| GBM | Glioblastoma |
| CKAP4 | Cytoskeleton-Associated Protein 4 |
| DKK1 | Dickkopf1 |
| PDAC | Pancreatic Ductal Adenocarcinoma |
| PD1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| GMP | Good Manufacturing Practice |
| EPR | Enhanced Permeability and Retention |
| PSC | Pancreatic Stellate Cell |
| MSC | Mesenchymal Stem Cells |
| HeLa | Henrietta Lacks Cells |
| PTX | Paclitaxel |
| KRAS | Kirsten Rat Sarcoma |
| DEX | Dendrite Cell- Derived Exosomes |
| NSCLC | Non-Small Cell Lung Cancer |
| MAGE-3 | Melanoma-Associated Antigen 3 |
| IFN-γ-free DEX | Interferon Gamma-free Exosomes |
| IFN-γ-DEX | Interferon Gamma-containing Exosomes |
| AEX | Ascites-Derived Exosomes |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| CTL | Cytotoxic T Lymphocyte |
| TH1 | T-cell Helper 1 |
| GNV | Grapefruit-Derived Nanovectors |
| PFS | Progression-Free Survival |
| OS | Overall Survival |
References
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [PubMed]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baj-Krzyworzeka, M.; Szatanek, R.; Weglarczyk, K.; Baran, J.; Urbanowicz, B.; Branski, P.; Ratajczak, M.Z.; Zembala, M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol. Immun. 2006, 55, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef]
- Aliotta, J.M.; Sanchez-Guijo, F.M.; Dooner, G.J.; Johnson, K.W.; Dooner, M.S.; Greer, K.A.; Greer, D.; Pimentel, J.; Kolankiewicz, L.M.; Puente, N.; et al. Alteration of marrow cell gene expression, protein production, and engraftment into lung by lung-derived microvesicles: A novel mechanism for phenotype modulation. Stem Cells 2007, 25, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Bonucci, E. Fine structure and histochemistry of “calcifying globules” in epiphyseal cartilage. Z. Zellforsch. Mikrosk. Anat. 1970, 103, 192–217. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Crescitelli, R.; Lasser, C.; Szabo, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzas, E.I.; Lotvall, J. Distinct RNA profiles in subpopulations of extracellular vesicles: Apoptotic bodies, microvesicles and exosomes. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borras, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. Plos Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Charoenviriyakul, C.; Takahashi, Y.; Morishita, M.; Matsumoto, A.; Nishikawa, M.; Takakura, Y. Cell type-specific and common characteristics of exosomes derived from mouse cell lines: Yield, physicochemical properties, and pharmacokinetics. Eur. J. Pharm. Sci. 2017, 96, 316–322. [Google Scholar] [CrossRef]
- Kim, D.K.; Kang, B.; Kim, O.Y.; Choi, D.S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C.; et al. EVpedia: An integrated database of high-throughput data for systemic analyses of extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Simpson, R.J.; Kalra, H.; Mathivanan, S. ExoCarta as a resource for exosomal research. J. Extracell. Vesicles 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannafon, B.N.; Ding, W.Q. Intercellular Communication by Exosome-Derived microRNAs in Cancer. Int. J. Mol. Sci. 2013, 14, 14240–14269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Hannafon, B.N.; Carpenter, K.J.; Berry, W.L.; Janknecht, R.; Dooley, W.C.; Ding, W.Q. Exosome-mediated microRNA signaling from breast cancer cells is altered by the anti-angiogenesis agent docosahexaenoic acid (DHA). Mol. Cancer 2015, 14, 133. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [Green Version]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [Green Version]
- Ismail, N.; Wang, Y.; Dakhlallah, D.; Moldovan, L.; Agarwal, K.; Batte, K.; Shah, P.; Wisler, J.; Eubank, T.D.; Tridandapani, S.; et al. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood 2013, 121, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, Y.; Chen, B.; Zhao, J.; Yu, S.; Tang, Y.; Zheng, Q.; Li, Y.; Wang, P.; He, X.; et al. exoRBase: A database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res. 2018, 46, D106–D112. [Google Scholar] [CrossRef] [Green Version]
- Murillo, O.D.; Thistlethwaite, W.; Rozowsky, J.; Subramanian, S.L.; Lucero, R.; Shah, N.; Jackson, A.R.; Srinivasan, S.; Chung, A.; Laurent, C.D.; et al. exRNA Atlas Analysis Reveals Distinct Extracellular RNA Cargo Types and Their Carriers Present across Human Biofluids. Cell 2019, 117, 463–477 e415. [Google Scholar] [CrossRef] [Green Version]
- Russo, F.; Di Bella, S.; Vannini, F.; Berti, G.; Scoyni, F.; Cook, H.V.; Santos, A.; Nigita, G.; Bonnici, V.; Lagana, A.; et al. miRandola 2017: A curated knowledge base of non-invasive biomarkers. Nucleic Acids Res. 2018, 46, D354–D359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnette, A.; Chatelain, M.; Chatelain, B.; Ten Cate, H.; Mullier, F. Pre-analytical issues in the haemostasis laboratory: Guidance for the clinical laboratories. Thromb. J. 2016, 14, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carayon, K.; Chaoui, K.; Ronzier, E.; Lazar, I.; Bertrand-Michel, J.; Roques, V.; Balor, S.; Terce, F.; Lopez, A.; Salome, L.; et al. Proteolipidic composition of exosomes changes during reticulocyte maturation. J. Biol. Chem. 2011, 286, 34426–34439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, O.; Schifferli, J.A. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 2004, 104, 2543–2548. [Google Scholar] [CrossRef]
- Montecalvo, A.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.G.; Wang, Z.; Divito, S.J.; Papworth, G.D.; Watkins, S.C.; Robbins, P.D.; Larregina, A.T.; et al. Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J. Immunol. 2008, 180, 3081–3090. [Google Scholar] [CrossRef] [Green Version]
- Mincheva-Nilsson, L.; Baranov, V. Placenta-derived exosomes and syncytiotrophoblast microparticles and their role in human reproduction: Immune modulation for pregnancy success. Am. J. Reprod. Immunol. 2014, 72, 440–457. [Google Scholar] [CrossRef]
- Lakkaraju, A.; Rodriguez-Boulan, E. Itinerant exosomes: Emerging roles in cell and tissue polarity. Trends Cell Biol. 2008, 18, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Quesenberry, P.J.; Aliotta, J.M. The paradoxical dynamism of marrow stem cells: Considerations of stem cells, niches, and microvesicles. Stem Cell Rev. 2008, 4, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Nahar, N.N.; Missana, L.R.; Garimella, R.; Tague, S.E.; Anderson, H.C. Matrix vesicles are carriers of bone morphogenetic proteins (BMPs), vascular endothelial growth factor (VEGF), and noncollagenous matrix proteins. J. Bone Min. Metab. 2008, 26, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Lee, S.A.; Clugston, R.D.; Blaner, W.S. Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 2012, 1821, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.P.; Breakefield, X.O. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front. Physiol. 2012, 3, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Chitiprolu, M.; Roncevic, L.; Javalet, C.; Hemming, F.J.; Trung, M.T.; Meng, L.; Latreille, E.; Tanese de Souza, C.; McCulloch, D.; et al. Atg5 Disassociates the V1V0-ATPase to Promote Exosome Production and Tumor Metastasis Independent of Canonical Macroautophagy. Dev. Cell 2017, 43, 716–730 e717. [Google Scholar] [CrossRef] [Green Version]
- Villarroya-Beltri, C.; Baixauli, F.; Mittelbrunn, M.; Fernandez-Delgado, I.; Torralba, D.; Moreno-Gonzalo, O.; Baldanta, S.; Enrich, C.; Guerra, S.; Sanchez-Madrid, F. ISGylation controls exosome secretion by promoting lysosomal degradation of MVB proteins. Nat. Commun. 2016, 7, 13588. [Google Scholar] [CrossRef] [Green Version]
- Ilie, A.; Gao, A.Y.L.; Boucher, A.; Park, J.; Berghuis, A.M.; Hoffer, M.J.V.; Hilhorst-Hofstee, Y.; McKinney, R.A.; Orlowski, J. A potential gain-of-function variant of SLC9A6 leads to endosomal alkalinization and neuronal atrophy associated with Christianson Syndrome. Neurobiol. Dis. 2019, 121, 187–204. [Google Scholar] [CrossRef]
- Edgar, J.R.; Manna, P.T.; Nishimura, S.; Banting, G.; Robinson, M.S. Tetherin is an exosomal tether. Elife 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Putz, U.; Howitt, J.; Lackovic, J.; Foot, N.; Kumar, S.; Silke, J.; Tan, S.S. Nedd4 family-interacting protein 1 (Ndfip1) is required for the exosomal secretion of Nedd4 family proteins. J. Biol. Chem. 2008, 283, 32621–32627. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.C. Molecular mechanisms of cellular cholesterol efflux. J. Biol. Chem. 2014, 289, 24020–24029. [Google Scholar] [CrossRef] [Green Version]
- Amzallag, N.; Passer, B.J.; Allanic, D.; Segura, E.; Thery, C.; Goud, B.; Amson, R.; Telerman, A. TSAP6 facilitates the secretion of translationally controlled tumor protein/histamine-releasing factor via a nonclassical pathway. J. Biol. Chem. 2004, 279, 46104–46112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xavier, C.P.R.; Caires, H.R.; Barbosa, M.A.G.; Bergantim, R.; Guimaraes, J.E.; Vasconcelos, M.H. The Role of Extracellular Vesicles in the Hallmarks of Cancer and Drug Resistance. Cells 2020, 9, 1141. [Google Scholar] [CrossRef]
- Antonyak, M.A.; Li, B.; Boroughs, L.K.; Johnson, J.L.; Druso, J.E.; Bryant, K.L.; Holowka, D.A.; Cerione, R.A. Cancer cell-derived microvesicles induce transformation by transferring tissue transglutaminase and fibronectin to recipient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4852–4857. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Vasaikar, S.; Eskaros, A.; Kim, Y.; Lewis, J.S.; Zhang, B.; Zijlstra, A.; Weaver, A.M. EPHB2 carried on small extracellular vesicles induces tumor angiogenesis via activation of ephrin reverse signaling. Jci Insight 2019, 4. [Google Scholar] [CrossRef]
- Bao, L.; You, B.; Shi, S.; Shan, Y.; Zhang, Q.; Yue, H.; Zhang, J.; Zhang, W.; Shi, Y.; Liu, Y.; et al. Metastasis-associated miR-23a from nasopharyngeal carcinoma-derived exosomes mediates angiogenesis by repressing a novel target gene TSGA10. Oncogene 2018, 37, 2873–2889. [Google Scholar] [CrossRef]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135 e110. [Google Scholar] [CrossRef] [Green Version]
- Setti, M.; Osti, D.; Richichi, C.; Ortensi, B.; Del Bene, M.; Fornasari, L.; Beznoussenko, G.; Mironov, A.; Rappa, G.; Cuomo, A.; et al. Extracellular vesicle-mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015, 6, 31413–31427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kgamma to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marleau, A.M.; Chen, C.S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shedden, K.; Xie, X.T.; Chandaroy, P.; Chang, Y.T.; Rosania, G.R. Expulsion of small molecules in vesicles shed by cancer cells: Association with gene expression and chemosensitivity profiles. Cancer Res. 2003, 63, 4331–4337. [Google Scholar]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427 e413. [Google Scholar] [CrossRef] [Green Version]
- Andaloussi, S.E.L.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Urabe, F.; Kosaka, N.; Ito, K.; Kimura, T.; Egawa, S.; Ochiya, T. Extracellular vesicles as biomarkers and therapeutic targets for cancer. Am. J. Physiol. Cell Physiol. 2020, 318, C29–C39. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kosaka, N.; Ochiya, T. Latest advances in extracellular vesicles: From bench to bedside. Sci. Technol. Adv. Mater. 2019, 20, 746–757. [Google Scholar] [CrossRef] [Green Version]
- D’Souza-Schorey, C.; Schorey, J.S. Regulation and mechanisms of extracellular vesicle biogenesis and secretion. Essays Biochem. 2018, 62, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30, sup pp. 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, R.C.; Rogers, R.G.; De Couto, G.; Tseliou, E.; Luther, K.; Holewinski, R.; Soetkamp, D.; Van Eyk, J.E.; Antes, T.J.; Marban, E. Newt cells secrete extracellular vesicles with therapeutic bioactivity in mammalian cardiomyocytes. J. Extracell. Vesicles 2018, 7, 1456888. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 2013, 288, 10849–10859. [Google Scholar] [CrossRef] [Green Version]
- Phuyal, S.; Hessvik, N.P.; Skotland, T.; Sandvig, K.; Llorente, A. Regulation of exosome release by glycosphingolipids and flotillins. Febs J. 2014, 281, 2214–2227. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [Green Version]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Thery, C. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef] [Green Version]
- Savina, A.; Fader, C.M.; Damiani, M.T.; Colombo, M.I. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 2005, 6, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Gronborg, M.; Mobius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Sturgeon, C.; Lamerz, R.; Haglund, C.; Holubec, V.L.; Klapdor, R.; Nicolini, A.; Topolcan, O.; Heinemann, V. Tumor markers in pancreatic cancer: A European Group on Tumor Markers (EGTM) status report. Ann. Oncol. 2010, 21, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [Green Version]
- Jahn, R.; Lang, T.; Sudhof, T.C. Membrane fusion. Cell 2003, 112, 519–533. [Google Scholar] [CrossRef] [Green Version]
- Gonda, A.; Kabagwira, J.; Senthil, G.N.; Wall, N.R. Internalization of Exosomes through Receptor-Mediated Endocytosis. Mol Cancer Res. 2019, 17, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Zhu, Y.L.; Zhou, Y.Y.; Liang, G.F.; Wang, Y.Y.; Hu, F.H.; Xiao, Z.D. Exosome uptake through clathrin-mediated endocytosis and macropinocytosis and mediating miR-21 delivery. J. Biol. Chem. 2014, 289, 22258–22267. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Zhao, W.L.; Ye, Y.Y.; Bai, X.C.; Liu, R.Q.; Chang, L.F.; Zhou, Q.; Sui, S.F. Cellular internalization of exosomes occurs through phagocytosis. Traffic 2010, 11, 675–687. [Google Scholar] [CrossRef]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, K.J.; Christianson, H.C.; Wittrup, A.; Bourseau-Guilmain, E.; Lindqvist, E.; Svensson, L.M.; Morgelin, M.; Belting, M. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J. Biol. Chem. 2013, 288, 17713–17724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. Bmc Cancer 2011, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Banizs, A.B.; Huang, T.; Nakamoto, R.K.; Shi, W.; He, J. Endocytosis Pathways of Endothelial Cell Derived Exosomes. Mol. Pharm. 2018, 15, 5585–5590. [Google Scholar] [CrossRef]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Tu, C.; Zhang, J.; Wang, J. Inhibition of multiple myelomaderived exosomes uptake suppresses the functional response in bone marrow stromal cell. Int. J. Oncol. 2019, 54, 1061–1070. [Google Scholar] [CrossRef]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’Connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Lentz, M.R. Continuous whole blood UltraPheresis procedure in patients with metastatic cancer. J. Biol. Response Mod. 1989, 8, 511–527. [Google Scholar]
- Tullis, R.H.; Duffin, R.P.; Handley, H.H.; Sodhi, P.; Menon, J.; Joyce, J.A.; Kher, V. Reduction of hepatitis C virus using lectin affinity plasmapheresis in dialysis patients. Blood Purif. 2009, 27, 64–69. [Google Scholar] [CrossRef]
- Tullis, R.H.; Duffin, R.P.; Zech, M.; Ambrus, J.L. Affinity hemodialysis for antiviral therapy. II. Removal of HIV-1 viral proteins from cell culture supernatants and whole blood. Blood Purif. 2003, 21, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Nie, H.; Zhou, Y.; Lian, S.; Mei, H.; Lu, Y.; Dong, H.; Li, F.; Li, T.; Li, B.; et al. Eliminating blood oncogenic exosomes into the small intestine with aptamer-functionalized nanoparticles. Nat. Commun. 2019, 10, 5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, H.; Yamamoto, H.; Harada, T.; Fumoto, K.; Osugi, Y.; Sada, R.; Maehara, N.; Hikita, H.; Mori, S.; Eguchi, H.; et al. CKAP4, a DKK1 Receptor, Is a Biomarker in Exosomes Derived from Pancreatic Cancer and a Molecular Target for Therapy. Clin. Cancer Res. 2019, 25, 1936–1947. [Google Scholar] [CrossRef] [Green Version]
- Binenbaum, Y.; Fridman, E.; Yaari, Z.; Milman, N.; Schroeder, A.; Ben David, G.; Shlomi, T.; Gil, Z. Transfer of miRNA in Macrophage-Derived Exosomes Induces Drug Resistance in Pancreatic Adenocarcinoma. Cancer Res. 2018, 78, 5287–5299. [Google Scholar] [CrossRef] [Green Version]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 Controls Exosome Synthesis and Promotes Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef]
- Li, X.J.; Ren, Z.J.; Tang, J.H.; Yu, Q. Exosomal MicroRNA MiR-1246 Promotes Cell Proliferation, Invasion and Drug Resistance by Targeting CCNG2 in Breast Cancer. Cell Physiol. Biochem. 2017, 44, 1741–1748. [Google Scholar] [CrossRef]
- Chen, J.H.; Wu, A.T.H.; Bamodu, O.A.; Yadav, V.K.; Chao, T.Y.; Tzeng, Y.M.; Mukhopadhyay, D.; Hsiao, M.; Lee, J.C. Ovatodiolide Suppresses Oral Cancer Malignancy by Down-Regulating Exosomal Mir-21/STAT3/beta-Catenin Cargo and Preventing Oncogenic Transformation of Normal Gingival Fibroblasts. Cancers 2019, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Hannafon, B.N.; Trigoso, Y.D.; Calloway, C.L.; Zhao, Y.D.; Lum, D.H.; Welm, A.L.; Zhao, Z.J.; Blick, K.E.; Dooley, W.C.; Ding, W.Q. Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res. 2016, 18, 90. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.F.; Hannafon, B.N.; Khatri, U.; Gin, A.; Ding, W.Q. The origin of exosomal miR-1246 in human cancer cells. Rna Biol. 2019, 16, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Hannafon, B.N.; Zhao, Y.D.; Postier, R.G.; Ding, W.Q. Plasma exosome miR-196a and miR-1246 are potential indicators of localized pancreatic cancer. Oncotarget 2017, 8, 77028–77040. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Page, D.B.; Postow, M.A.; Callahan, M.K.; Allison, J.P.; Wolchok, J.D. Immune modulation in cancer with antibodies. Annu. Rev. Med. 2014, 65, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fan, Y.; Che, X.; Zhang, M.; Li, Z.; Li, C.; Wang, S.; Wen, T.; Hou, K.; Shao, X.; et al. Anti-PD-1 Therapy Response Predicted by the Combination of Exosomal PD-L1 and CD28. Front. Oncol. 2020, 10, 760. [Google Scholar] [CrossRef]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Wang, W.; Chen, R.; Zhang, Y.; Zou, K.; Ye, M.; He, X.; Zhang, F.; Han, J. Exosome-mediated transfer of lncRNASNHG14 promotes trastuzumab chemoresistance in breast cancer. Int. J. Oncol. 2018, 53, 1013–1026. [Google Scholar] [CrossRef] [Green Version]
- Milman, N.; Ginini, L.; Gil, Z. Exosomes and their role in tumorigenesis and anticancer drug resistance. Drug Resist. Updat 2019, 45, 1–12. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, in press. [Google Scholar] [CrossRef]
- Bastos, N.; Ruivo, C.F.; da Silva, S.; Melo, S.A. Exosomes in cancer: Use them or target them? Semin Cell Dev. Biol. 2018, 78, 13–21. [Google Scholar] [CrossRef]
- Ohno, S.; Drummen, G.P.; Kuroda, M. Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems. Int. J. Mol. Sci. 2016, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Smyth, T.J.; Redzic, J.S.; Graner, M.W.; Anchordoquy, T.J. Examination of the specificity of tumor cell derived exosomes with tumor cells in vitro. Biochim. Biophys. Acta 2014, 1838, 2954–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Zeng, Q.; Han, Q.; Xia, W. Effect of pH, temperature and freezing-thawing on quantity changes and cellular uptake of exosomes. Protein. Cell 2019, 10, 295–299. [Google Scholar] [CrossRef] [Green Version]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharm. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical Cancer Nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Tsibouklis, J.; Weng, T.; Zhang, B.; Yin, G.; Feng, G.; Cui, Y.; Savina, I.N.; Mikhalovska, L.I.; Sandeman, S.R.; et al. Nano carriers for drug transport across the blood-brain barrier. J. Drug Target. 2017, 25, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchanapally, R.; Deshmukh, S.K.; Chavva, S.R.; Tyagi, N.; Srivastava, S.K.; Patel, G.K.; Singh, A.P.; Singh, S. Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: A comparative analysis. Int. J. Nanomed. 2019, 14, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrelli, D.A.; Yankson, K.; Shukla, N.; Vilanilam, G.; Ticer, T.; Wolfram, J. Extracellular vesicle therapeutics for liver disease. J. Control. Release 2018, 273, 86–98. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Kordelas, L.; Rebmann, V.; Ludwig, A.K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Munoz, J.L.; Bliss, S.A.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Delivery of Functional Anti-miR-9 by Mesenchymal Stem Cell-derived Exosomes to Glioblastoma Multiforme Cells Conferred Chemosensitivity. Mol. Nucleic Acids 2013, 2, e126. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, L.; Cocce, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Vigano, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Lohan, P.; Treacy, O.; Griffin, M.D.; Ritter, T.; Ryan, A.E. Anti-Donor Immune Responses Elicited by Allogeneic Mesenchymal Stem Cells and Their Extracellular Vesicles: Are We Still Learning? Front. Immunol. 2017, 8, 1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpinen, L.; Impola, U.; Sankkila, L.; Ritamo, I.; Aatonen, M.; Kilpinen, S.; Tuimala, J.; Valmu, L.; Levijoki, J.; Finckenberg, P.; et al. Extracellular membrane vesicles from umbilical cord blood-derived MSC protect against ischemic acute kidney injury, a feature that is lost after inflammatory conditioning. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Kharaziha, P.; Ceder, S.; Li, Q.; Panaretakis, T. Tumor cell-derived exosomes: A message in a bottle. Biochim. Biophys. Acta 2012, 1826, 103–111. [Google Scholar] [CrossRef]
- Shtam, T.A.; Kovalev, R.A.; Varfolomeeva, E.Y.; Makarov, E.M.; Kil, Y.V.; Filatov, M.V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun. Signal. 2013, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Lou, G.; Song, X.; Yang, F.; Wu, S.; Wang, J.; Chen, Z.; Liu, Y. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J. Hematol. Oncol. 2015, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Abels, E.R.; Maas, S.L.N.; Nieland, L.; Wei, Z.; Cheah, P.S.; Tai, E.; Kolsteeg, C.J.; Dusoswa, S.A.; Ting, D.T.; Hickman, S.; et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep. 2019, 28, 3105–3119 e3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fareh, M.; Almairac, F.; Turchi, L.; Burel-Vandenbos, F.; Paquis, P.; Fontaine, D.; Lacas-Gervais, S.; Junier, M.P.; Chneiweiss, H.; Virolle, T. Cell-based therapy using miR-302-367 expressing cells represses glioblastoma growth. Cell Death Dis. 2017, 8, e2713. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef]
- O’Loughlin, A.J.; Mager, I.; de Jong, O.G.; Varela, M.A.; Schiffelers, R.M.; El Andaloussi, S.; Wood, M.J.A.; Vader, P. Functional Delivery of Lipid-Conjugated siRNA by Extracellular Vesicles. Mol. Ther. 2017, 25, 1580–1587. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Amreddy, N.; Pareek, V.; Chinnappan, M.; Ahmed, R.; Mehta, M.; Razaq, M.; Munshi, A.; Ramesh, R. Progress in extracellular vesicle biology and their application in cancer medicine. Wiley Interdiscip Rev. Nanomed. Nanobiotechnol. 2020, 12, e1621. [Google Scholar] [CrossRef] [Green Version]
- Potter, H. Transfection by electroporation. Curr. Protoc. Mol. Biol. 2003. [Google Scholar] [CrossRef]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef] [Green Version]
- Hood, J.L.; Scott, M.J.; Wickline, S.A. Maximizing exosome colloidal stability following electroporation. Anal. Biochem. 2014, 448, 41–49. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Su, C. Design strategies and application progress of therapeutic exosomes. Theranostics 2019, 9, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Asadirad, A.; Hashemi, S.M.; Baghaei, K.; Ghanbarian, H.; Mortaz, E.; Zali, M.R.; Amani, D. Phenotypical and functional evaluation of dendritic cells after exosomal delivery of miRNA-155. Life Sci. 2019, 219, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wu, J.Y.; Wang, J.M.; Hu, X.B.; Cai, J.X.; Xiang, D.X. Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomater. 2020, 101, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.A.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Goh, W.J.; Lee, C.K.; Zou, S.; Woon, E.C.; Czarny, B.; Pastorin, G. Doxorubicin-loaded cell-derived nanovesicles: An alternative targeted approach for anti-tumor therapy. Int. J. Nanomed. 2017, 12, 2759–2767. [Google Scholar] [CrossRef] [Green Version]
- Kalimuthu, S.; Gangadaran, P.; Rajendran, R.L.; Zhu, L.; Oh, J.M.; Lee, H.W.; Gopal, A.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; et al. A New Approach for Loading Anticancer Drugs Into Mesenchymal Stem Cell-Derived Exosome Mimetics for Cancer Therapy. Front. Pharm. 2018, 9, 1116. [Google Scholar] [CrossRef]
- Bunggulawa, E.J.; Wang, W.; Yin, T.; Wang, N.; Durkan, C.; Wang, Y.; Wang, G. Recent advancements in the use of exosomes as drug delivery systems. J. Nanobiotechnol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbarghazi, R.; Jabbari, N.; Sani, N.A.; Asghari, R.; Salimi, L.; Kalashani, S.A.; Feghhi, M.; Etemadi, T.; Akbariazar, E.; Mahmoudi, M.; et al. Tumor-derived extracellular vesicles: Reliable tools for Cancer diagnosis and clinical applications. Cell Commun. Signal. 2019, 17, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.F.; Shi, J.B.; Li, C. Small extracellular vesicle loading systems in cancer therapy: Current status and the way forward. Cytotherapy 2019, 21, 1122–1136. [Google Scholar] [CrossRef]
- Walker, S.; Busatto, S.; Pham, A.; Tian, M.; Suh, A.; Carson, K.; Quintero, A.; Lafrence, M.; Malik, H.; Santana, M.X.; et al. Extracellular vesicle-based drug delivery systems for cancer treatment. Theranostics 2019, 9, 8001–8017. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Lin, E.Y.; Chiou, T.W.; Harn, H.J. Exosomes in clinical trial and their production in compliance with good manufacturing practice. Tzu-Chi Med. J. 2019, 32, 113–120. [Google Scholar] [CrossRef]
- Guo, M.; Wu, F.; Hu, G.; Chen, L.; Xu, J.; Xu, P.; Wang, X.; Li, Y.; Liu, S.; Zhang, S.; et al. Autologous tumor cell-derived microparticle-based targeted chemotherapy in lung cancer patients with malignant pleural effusion. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Zhang, H.; Xu, P.; Liu, J.; Ma, J.; Lv, M.; Li, D.; Katirai, F.; Shen, G.X.; et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 2012, 3, 1282. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhang, Y.; Tang, K.; Zhang, H.; Yin, X.; Li, Y.; Xu, P.; Sun, Y.; Ma, R.; Ji, T.; et al. Reversing drug resistance of soft tumor-repopulating cells by tumor cell-derived chemotherapeutic microparticles. Cell Res. 2016, 26, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Somiya, M.; Yoshioka, Y.; Ochiya, T. Drug delivery application of extracellular vesicles; insight into production, drug loading, targeting, and pharmacokinetics. Aims Bioeng. 2017, 4, 73–92. [Google Scholar] [CrossRef]
- Wang, Q.; Zhuang, X.; Mu, J.; Deng, Z.B.; Jiang, H.; Zhang, L.; Xiang, X.; Wang, B.; Yan, J.; Miller, D.; et al. Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids. Nat. Commun. 2013, 4, 1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnouf, T.; Agrahari, V.; Agrahari, V. Extracellular Vesicles As Nanomedicine: Hopes And Hurdles In Clinical Translation. Int. J. Nanomed. 2019, 14, 8847–8859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrahari, V.; Agrahari, V.; Burnouf, P.A.; Chew, C.H.; Burnouf, T. Extracellular Microvesicles as New Industrial Therapeutic Frontiers. Trends Biotechnol. 2019, 37, 707–729. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Disease | Drug | EV Source | Phase, n of Patients | Status | Reference |
|---|---|---|---|---|---|
| Malignant Pleural Effusion | Methotrexate | Autologous Tumor-Derived Microparticles | Phase 2, n = 90 | Recruiting | NCT02657460 1 Guo, M. [163] |
| Methotrexate | Microparticles | N/A, n = 248 | Recruiting | NCT04131231 1 | |
| Chemotherapeutic Drugs | Tumor Cell- Derived Microparticles | Phase 2, n = 30 | Unknown | NCT01854866 1 Tang, K. [164] | |
| Cisplatin | Tumor Cell- Derived Microparticles | N/A, n = 6 | Completed | Ma, J. [165] | |
| Metastatic Pancreatic Cancer | KRAS 2 G12D siRNA | MSC 3-Derived Exosomes | Phase 1, n = 28 | Recruiting | NCT03608631 1 Kamerkar, S. [166] |
| Head and Neck Cancer | Grape Extract | Plant Exosomes | Phase 1, n = 60 | Active, Not Recruiting | NCT01668849 1 |
| Hemopurifier Pembro-lizumab | Blood-Derived Exosomes | N/A, n = 12 | Not Yet Recruiting | NCT04453046 1 | |
| Colorectal Cancer | Curcumin | Plant Exosomes | Phase 1, n = 7 | Active, Not Recruiting | NCT01294072 1 |
| GM-CSF 4 | AEX 5 | Phase 1, n = 40 | Completed | Dai, S. [167] | |
| Non-Small Cell Lung Cancer | Antigens | Tumor Dex2 6 | Phase 2, n = 41 | Completed | NCT01159288 1 Besse, B. [168] |
| MAGE7 Tumor Antigens | Autologous DEX 6 | Phase 1, n = 13 | Completed | Morse, M.A. [169] | |
| Metastatic Melanoma | MAGE7 3 Peptides | Autologous DEX 6 | Phase 1, n = 15 | Completed | Escudier, B. [170] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Burrola, S.; Wu, J.; Ding, W.-Q. Extracellular Vesicles in the Development of Cancer Therapeutics. Int. J. Mol. Sci. 2020, 21, 6097. https://doi.org/10.3390/ijms21176097
Sun H, Burrola S, Wu J, Ding W-Q. Extracellular Vesicles in the Development of Cancer Therapeutics. International Journal of Molecular Sciences. 2020; 21(17):6097. https://doi.org/10.3390/ijms21176097
Chicago/Turabian StyleSun, Haoyao, Stephanie Burrola, Jinchang Wu, and Wei-Qun Ding. 2020. "Extracellular Vesicles in the Development of Cancer Therapeutics" International Journal of Molecular Sciences 21, no. 17: 6097. https://doi.org/10.3390/ijms21176097
APA StyleSun, H., Burrola, S., Wu, J., & Ding, W. -Q. (2020). Extracellular Vesicles in the Development of Cancer Therapeutics. International Journal of Molecular Sciences, 21(17), 6097. https://doi.org/10.3390/ijms21176097