Lamin A/C and the Immune System: One Intermediate Filament, Many Faces



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Lamin A/C
2. Immune System
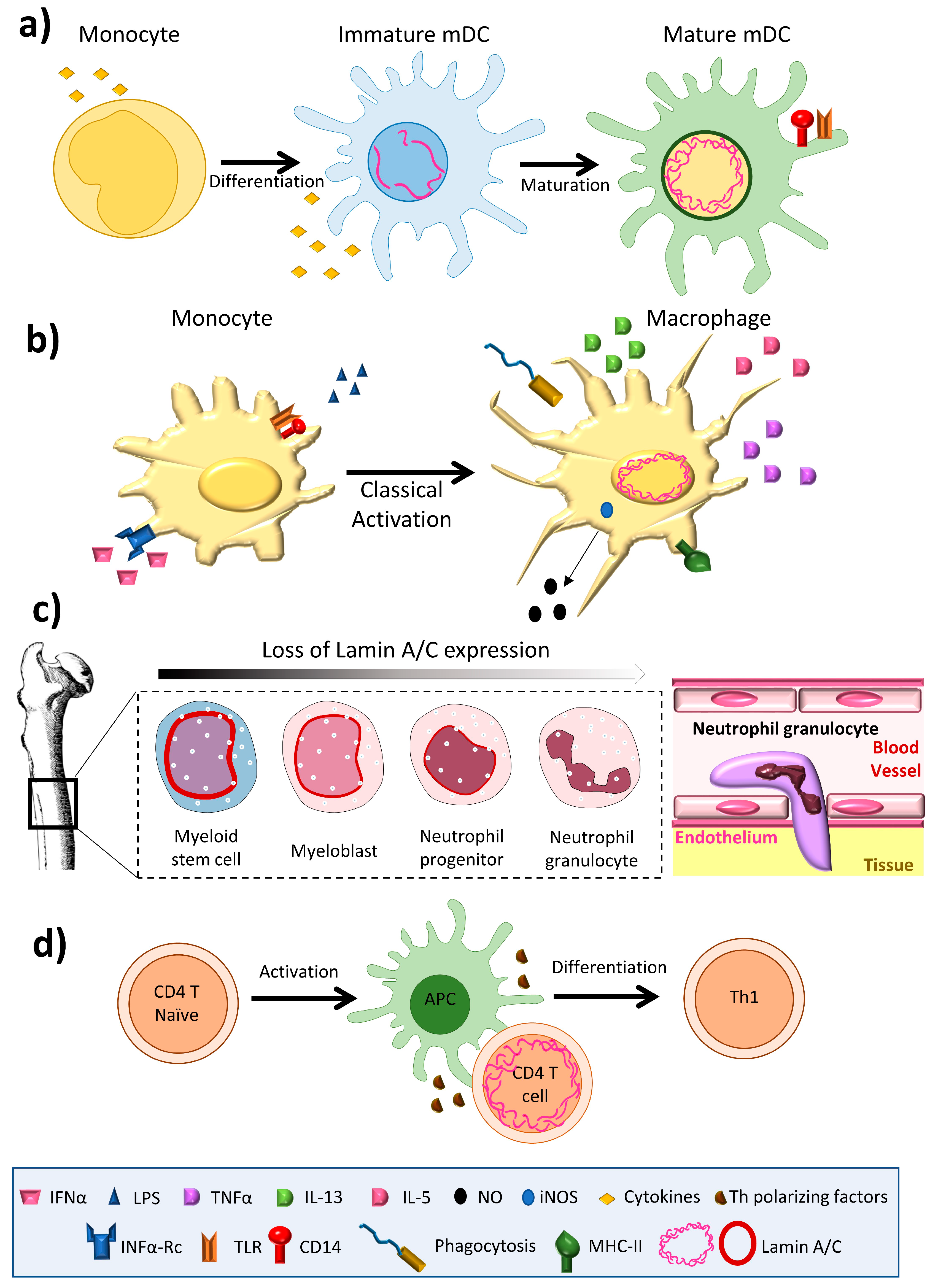
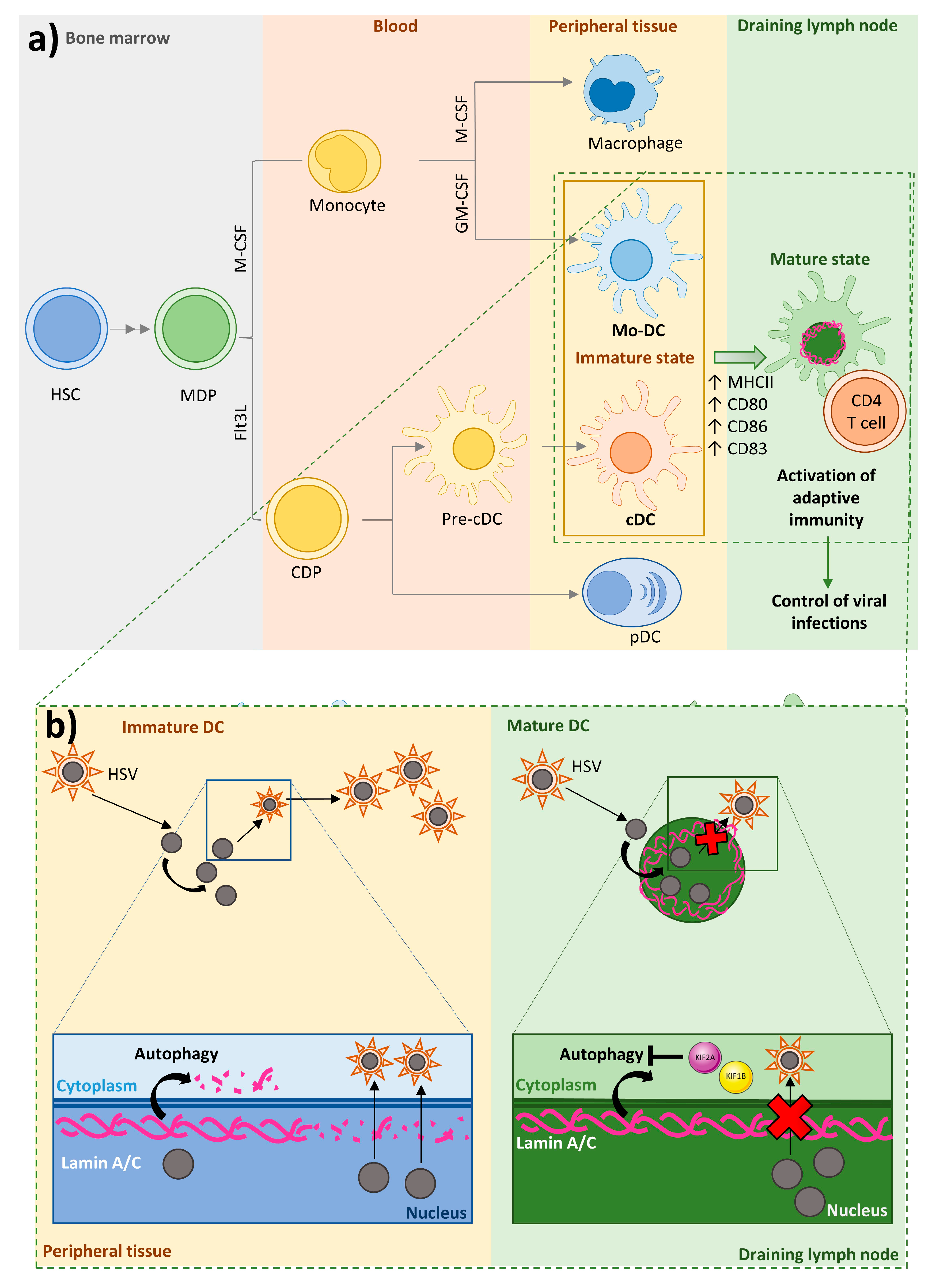
3. Lamin A/C Expression in Immune Cells
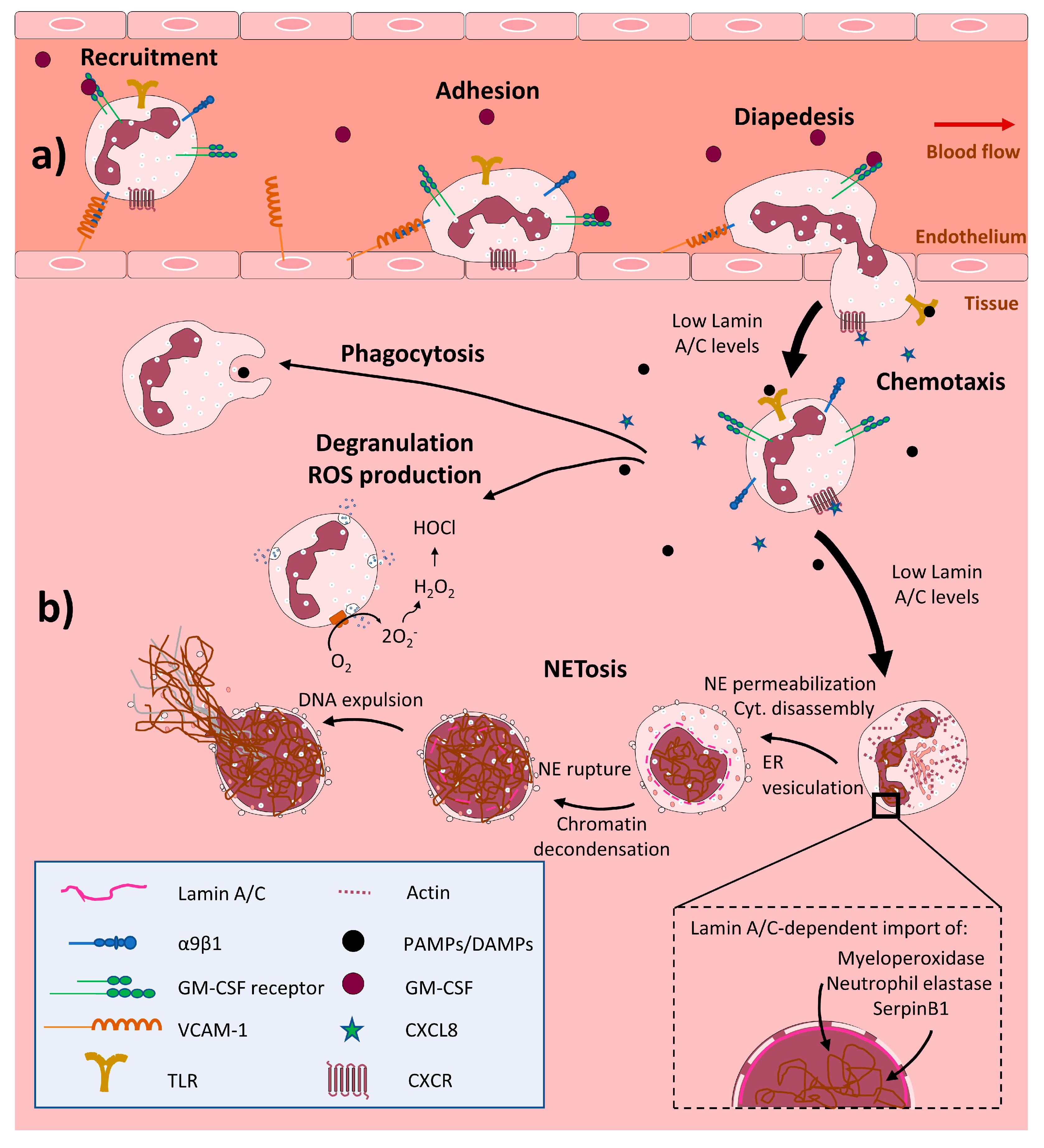
4. Lamin A/C in Innate Immunity
4.1. Neutrophils
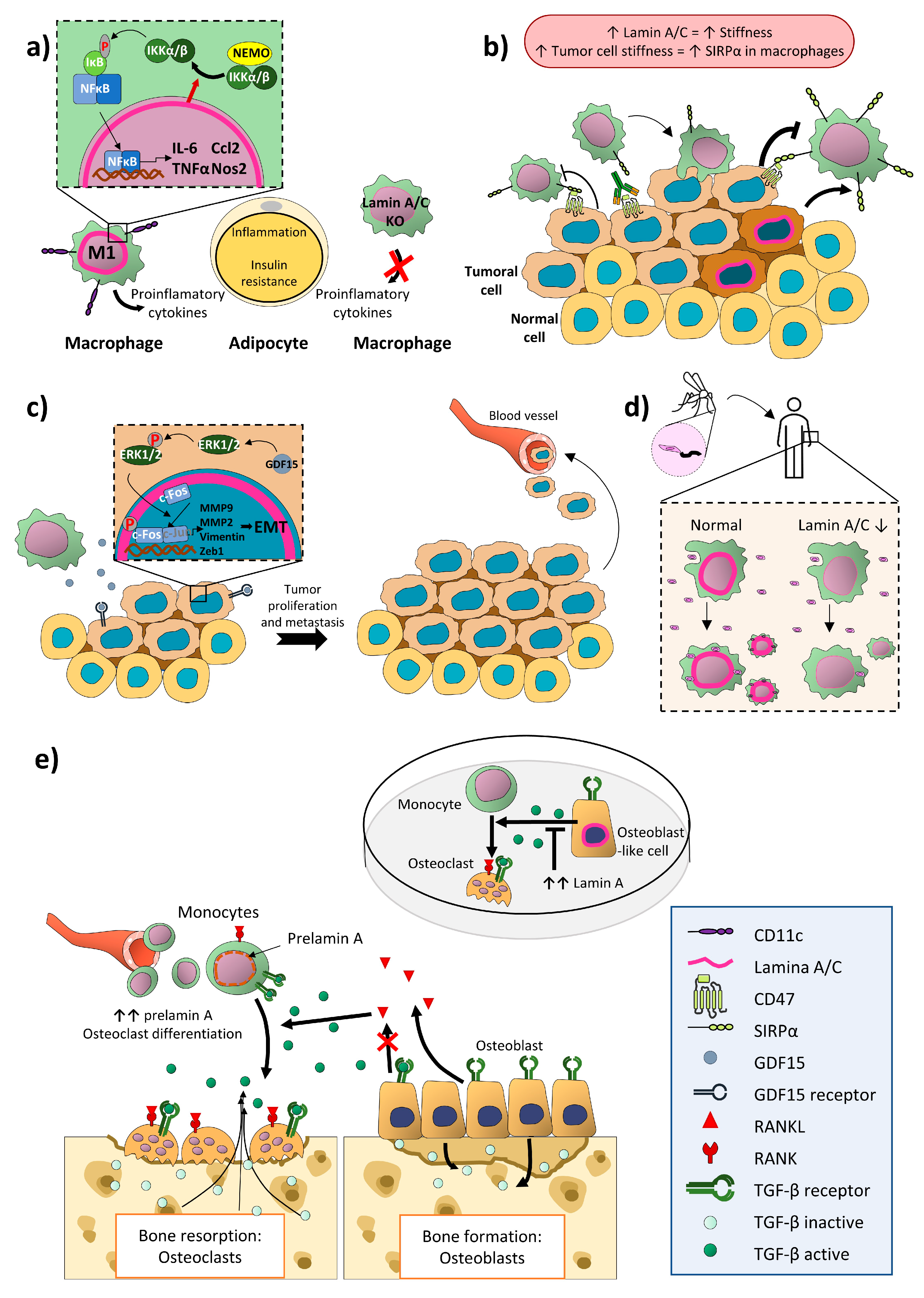
4.2. Monocytes/Macrophages
4.3. Dendritic Cells
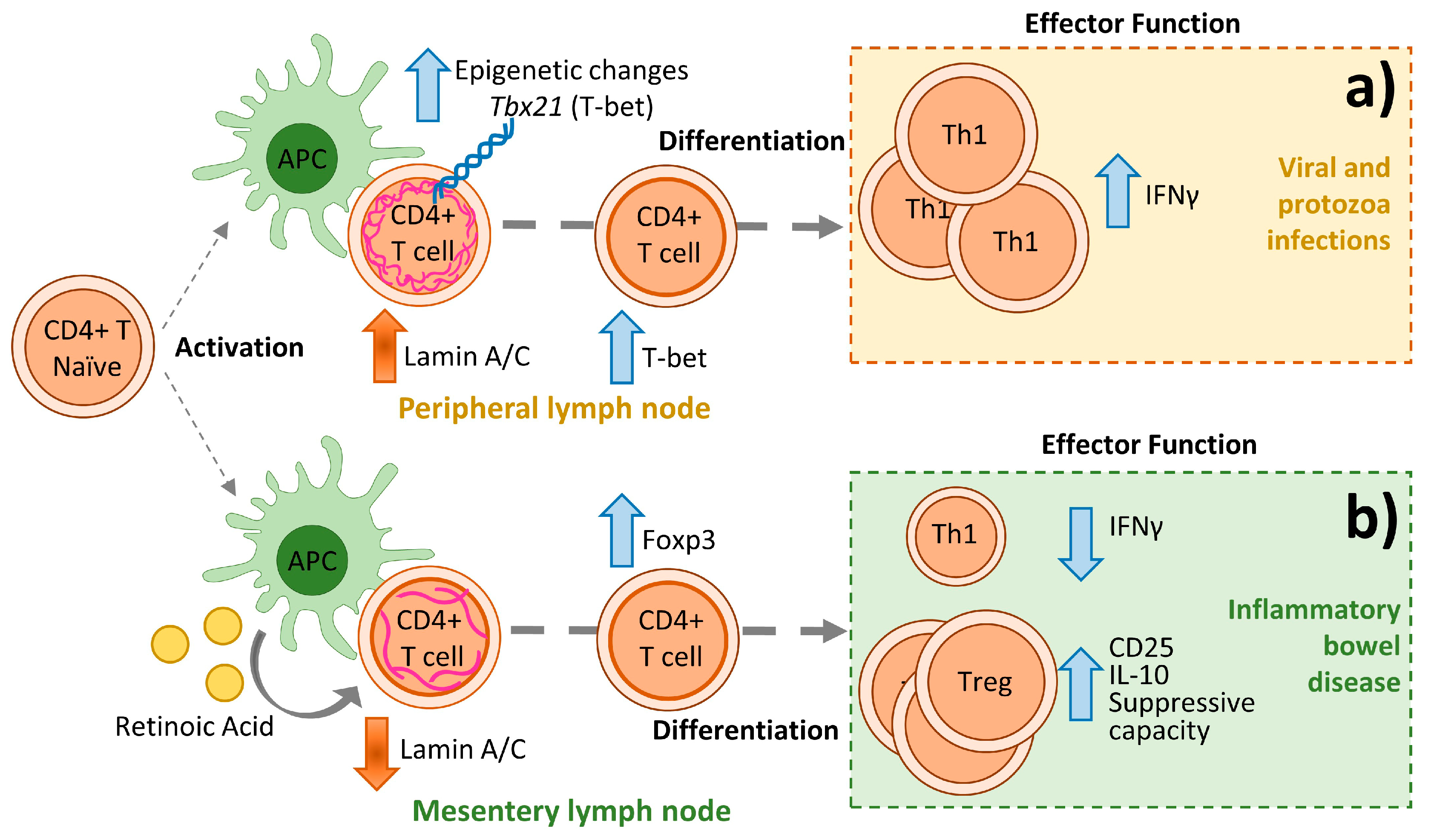
5. Lamin A/C in Adaptive Immunity
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | activator protein 1 |
| CCL | chemokine (C-C motif) ligand |
| COPD | chronic obstructive pulmonary disease |
| CSF-1 | colony-stimulating factor-1 |
| cSMAC | central supramolecular activation cluster |
| CX3CL | chemokine (C-X3-C motif) ligand |
| CXCL | chemokine (C-X-C motif) ligand |
| DAMPs | damage-associated molecular patterns |
| DCs | dendritic cells |
| GDF15 | growth differentiation factor 15 |
| HA | hyaluronic acid |
| HIV-1 | human immunodeficiency virus type 1 |
| HL-60 | human promyelocytic leukemia cells |
| HSV-1 | herpex Simplex Virus |
| LmnaDhe | hair and ears allele Lmna |
| LPS | lipopolysaccharide |
| MHC-I | antigen/major histocompatibility complex I |
| MHC-II | antigen/major histocompatibility complex II |
| MTOC | microtubule organizing center |
| NETs | neutrophil extracellular traps |
| NKT | cells natural killer T cells |
| NO | nitric oxide |
| PAMPs | pathogen-associated molecular patterns |
| PRRs | pattern recognition receptors |
| pSMAC | peripheral supramolecular activation cluster |
| RA | retinoic acid |
| ROS | reactive oxygen species |
| SUN2 | Sad1 and UNC84 domain containing 2 |
| TLRs | toll-like receptors |
| TPA | phorbol ester |
| TGF-β | transforming growth factor-β |
| VEGF | growth factors like vascular endothelial growth factor |
| Vpr | viral protein R |
References
- Broers, J.L.; Ramaekers, F.C.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.M.; Pla, D.; Perez-Sala, D.; Andres, V. A-type lamins and Hutchinson-Gilford progeria syndrome: Pathogenesis and therapy. Front. Biosci. (Schol. Ed.) 2011, 3, 1133–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Adam, S.A.; Taimen, P.; Shimi, T.; Goldman, R.D. Nuclear lamins. Cold Spring Harb. Perspect. Biol. 2010, 2, a000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moir, R.D.; Yoon, M.; Khuon, S.; Goldman, R.D. Nuclear lamins A and B1: Different pathways of assembly during nuclear envelope formation in living cells. J. Cell Biol. 2000, 151, 1155–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andres, V.; Gonzalez, J.M. Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 2009, 187, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, E.C.; Foisner, R. Proteins that associate with lamins: Many faces, many functions. Exp. Cell Res. 2007, 313, 2167–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigueros-Motos, L.; Gonzalez, J.M.; Rivera, J.; Andres, V. Hutchinson-Gilford progeria syndrome, cardiovascular disease and oxidative stress. Front. Biosci. (Schol. Ed.) 2011, 3, 1285–1297. [Google Scholar] [CrossRef] [Green Version]
- Bassler, K.; Schulte-Schrepping, J.; Warnat-Herresthal, S.; Aschenbrenner, A.C.; Schultze, J.L. The Myeloid Cell Compartment-Cell by Cell. Annu. Rev. Immunol. 2019, 37, 269–293. [Google Scholar] [CrossRef]
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [Green Version]
- Ruterbusch, M.; Pruner, K.B.; Shehata, L.; Pepper, M. In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu. Rev. Immunol. 2020, 38, 705–725. [Google Scholar] [CrossRef]
- Eckersley-Maslin, M.A.; Bergmann, J.H.; Lazar, Z.; Spector, D.L. Lamin A/C is expressed in pluripotent mouse embryonic stem cells. Nucleus 2013, 4, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinescu, D.; Gray, H.L.; Sammak, P.J.; Schatten, G.P.; Csoka, A.B. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem. Cells 2006, 24, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Duque, G.; Rivas, D. Age-related changes in lamin A/C expression in the osteoarticular system: Laminopathies as a potential new aging mechanism. Mech. Ageing Dev. 2006, 127, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, R.K.; Rober, R.A.; Kuhn, R.; Weber, K.; Osborn, M.; Peters, J.H. Dendritic accessory cells derived from rat bone marrow precursors under chemically defined conditions in vitro belong to the myeloid lineage. Eur. J. Cell Biol. 1991, 54, 171–181. [Google Scholar]
- Gieseler, R.K.; Xu, H.; Schlemminger, R.; Peters, J.H. Serum-free differentiation of rat and human dendritic cells, accompanied by acquisition of the nuclear lamins A/C as differentiation markers. Adv. Exp. Med. Biol. 1993, 329, 287–291. [Google Scholar]
- Gonzalez-Granado, J.M.; Silvestre-Roig, C.; Rocha-Perugini, V.; Trigueros-Motos, L.; Cibrian, D.; Morlino, G.; Blanco-Berrocal, M.; Osorio, F.G.; Freije, J.M.P.; Lopez-Otin, C.; et al. Nuclear envelope lamin-A couples actin dynamics with immunological synapse architecture and T cell activation. Sci. Signal 2014, 7, ra37. [Google Scholar] [CrossRef] [Green Version]
- Contu, F.; Rangel-Pozzo, A.; Trokajlo, P.; Wark, L.; Klewes, L.; Johnson, N.A.; Petrogiannis-Haliotis, T.; Gartner, J.G.; Garini, Y.; Vanni, R.; et al. Distinct 3D Structural Patterns of Lamin A/C Expression in Hodgkin and Reed-Sternberg Cells. Cancers 2018, 10, 286. [Google Scholar] [CrossRef] [Green Version]
- Yabuki, M.; Miyake, T.; Doi, Y.; Fujiwara, T.; Hamazaki, K.; Yoshioka, T.; Horton, A.A.; Utsumi, K. Role of nuclear lamins in nuclear segmentation of human neutrophils. Physiol. Chem. Phys. Med. NMR 1999, 31, 77–84. [Google Scholar]
- Rocha-Perugini, V.; Gonzalez-Granado, J.M. Nuclear envelope lamin-A as a coordinator of T cell activation. Nucleus 2014, 5, 396–401. [Google Scholar] [CrossRef] [Green Version]
- Prechtel, A.T.; Turza, N.M.; Theodoridis, A.A.; Kummer, M.; Steinkasserer, A. Small interfering RNA (siRNA) delivery into monocyte-derived dendritic cells by electroporation. J. Immunol. Methods 2006, 311, 139–152. [Google Scholar] [CrossRef]
- Rober, R.A.; Gieseler, R.K.; Peters, J.H.; Weber, K.; Osborn, M. Induction of nuclear lamins A/C in macrophages in in vitro cultures of rat bone marrow precursor cells and human blood monocytes, and in macrophages elicited in vivo by thioglycollate stimulation. Exp. Cell Res. 1990, 190, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.H.; Ruppert, J.; Gieseler, R.K.; Najar, H.M.; Xu, H. Differentiation of human monocytes into CD14 negative accessory cells: Do dendritic cells derive from the monocytic lineage? Pathobiology 1991, 59, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Collard, J.F.; Senecal, J.L.; Raymond, Y. Redistribution of nuclear lamin A is an early event associated with differentiation of human promyelocytic leukemia HL-60 cells. J. Cell Sci. 1992, 101, 657–670. [Google Scholar] [PubMed]
- Jin, D.; Ojcius, D.M.; Sun, D.; Dong, H.; Luo, Y.; Mao, Y.; Yan, J. Leptospira interrogans induces apoptosis in macrophages via caspase-8- and caspase-3-dependent pathways. Infect. Immun. 2009, 77, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H. Expression of nuclear envelope lamins A and C in human myeloid leukemias. Cancer Res. 1992, 52, 2847–2853. [Google Scholar]
- Paulin-Levasseur, M.; Julien, M. Expression of intermediate filament proteins in TPA-induced MPC-11 and HL-60 cells. Exp. Cell Res. 1992, 199, 363–372. [Google Scholar] [CrossRef]
- Manley, H.R.; Keightley, M.C.; Lieschke, G.J. The Neutrophil Nucleus: An Important Influence on Neutrophil Migration and Function. Front. Immunol. 2018, 9, 2867. [Google Scholar] [CrossRef]
- Olins, A.L.; Zwerger, M.; Herrmann, H.; Zentgraf, H.; Simon, A.J.; Monestier, M.; Olins, D.E. The human granulocyte nucleus: Unusual nuclear envelope and heterochromatin composition. Eur. J. Cell Biol. 2008, 87, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Olins, A.L.; Ernst, A.; Zwerger, M.; Herrmann, H.; Olins, D.E. An in vitro model for Pelger-Huet anomaly: Stable knockdown of lamin B receptor in HL-60 cells. Nucleus 2010, 1, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Olins, A.L.; Herrmann, H.; Lichter, P.; Kratzmeier, M.; Doenecke, D.; Olins, D.E. Nuclear envelope and chromatin compositional differences comparing undifferentiated and retinoic acid- and phorbol ester-treated HL-60 cells. Exp. Cell Res. 2001, 268, 115–127. [Google Scholar] [CrossRef]
- Olins, A.L.; Hoang, T.V.; Zwerger, M.; Herrmann, H.; Zentgraf, H.; Noegel, A.A.; Karakesisoglou, I.; Hodzic, D.; Olins, D.E. The LINC-less granulocyte nucleus. Eur. J. Cell Biol. 2009, 88, 203–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiam, H.R.; Vargas, P.; Carpi, N.; Crespo, C.L.; Raab, M.; Terriac, E.; King, M.C.; Jacobelli, J.; Alberts, A.S.; Stradal, T.; et al. Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat. Commun. 2016, 7, 10997. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.P.; Machiels, B.M.; Hopman, A.H.; Broers, J.L.; Bot, F.J.; Arends, J.W.; Ramaekers, F.C.; Schouten, H.C. Comparison of A and B-type lamin expression in reactive lymph nodes and nodular sclerosing Hodgkin’s disease. Histopathology 1997, 31, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Guilly, M.N.; Bensussan, A.; Bourge, J.F.; Bornens, M.; Courvalin, J.C. A human T lymphoblastic cell line lacks lamins A and C. EMBO J. 1987, 6, 3795–3799. [Google Scholar] [CrossRef]
- Rober, R.A.; Sauter, H.; Weber, K.; Osborn, M. Cells of the cellular immune and hemopoietic system of the mouse lack lamins A/C: Distinction versus other somatic cells. J. Cell Sci. 1990, 95, 587–598. [Google Scholar]
- Stadelmann, B.; Khandjian, E.; Hirt, A.; Luthy, A.; Weil, R.; Wagner, H.P. Repression of nuclear lamin A and C gene expression in human acute lymphoblastic leukemia and non-Hodgkin’s lymphoma cells. Leuk Res. 1990, 14, 815–821. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Trifonova, R.; Vrbanac, V.; Basar, E.; McKernan, S.; Xu, Z.; Seung, E.; Deruaz, M.; Dudek, T.; Einarsson, J.I.; et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J. Clin. Investig. 2011, 121, 2401–2412. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.R.; Meier, R.; Hirt, A.; Bodmer, J.J.; Janic, D.; Leibundgut, K.; Luthy, A.R.; Wagner, H.P. Nuclear lamin expression reveals a surprisingly high growth fraction in childhood acute lymphoblastic leukemia cells. Leukemia 1994, 8, 940–945. [Google Scholar]
- Bauer, B.; Baier, G. Protein kinase C and AKT/protein kinase B in CD4+ T-lymphocytes: New partners in TCR/CD28 signal integration. Mol. Immunol. 2002, 38, 1087–1099. [Google Scholar] [CrossRef]
- Bertacchini, J.; Beretti, F.; Cenni, V.; Guida, M.; Gibellini, F.; Mediani, L.; Marin, O.; Maraldi, N.M.; de Pol, A.; Lattanzi, G.; et al. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J. 2013, 27, 2145–2155. [Google Scholar] [CrossRef]
- Zuo, B.; Yang, J.; Wang, F.; Wang, L.; Yin, Y.; Dan, J.; Liu, N.; Liu, L. Influences of lamin A levels on induction of pluripotent stem cells. Biol. Open 2012, 1, 1118–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, K.; Nakamachi, K.; Hosoe, Y.; Nakajima, N. Identification of a novel retinoic acid-responsive element within the lamin A/C promoter. Biochem. Biophys. Res. Commun. 2000, 269, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.W.; Spinler, K.R.; Swift, J.; Chasis, J.A.; Mohandas, N.; Discher, D.E. Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc. Natl. Acad. Sci. USA 2013, 110, 18892–18897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toribio-Fernandez, R.; Herrero-Fernandez, B.; Zorita, V.; Lopez, J.A.; Vazquez, J.; Criado, G.; Pablos, J.L.; Collas, P.; Sanchez-Madrid, F.; Andres, V.; et al. Lamin A/C deficiency in CD4(+) T-cells enhances regulatory T-cells and prevents inflammatory bowel disease. J. Pathol. 2019, 249, 509–522. [Google Scholar] [CrossRef]
- Pino-Lagos, K.; Benson, M.J.; Noelle, R.J. Retinoic acid in the immune system. Ann. N. Y. Acad. Sci. 2008, 1143, 170–187. [Google Scholar] [CrossRef] [Green Version]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef]
- Esterhazy, D.; Loschko, J.; London, M.; Jove, V.; Oliveira, T.Y.; Mucida, D. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T(reg) cells and tolerance. Nat. Immunol. 2016, 17, 545–555. [Google Scholar] [CrossRef]
- Brostjan, C.; Oehler, R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov. 2020, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Hellebrekers, P.; Vrisekoop, N.; Koenderman, L. Neutrophil phenotypes in health and disease. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12943. [Google Scholar] [CrossRef]
- Leliefeld, P.H.; Wessels, C.M.; Leenen, L.P.; Koenderman, L.; Pillay, J. The role of neutrophils in immune dysfunction during severe inflammation. Crit. Care 2016, 20, 73. [Google Scholar] [CrossRef] [Green Version]
- Juss, J.K.; House, D.; Amour, A.; Begg, M.; Herre, J.; Storisteanu, D.M.; Hoenderdos, K.; Bradley, G.; Lennon, M.; Summers, C.; et al. Acute Respiratory Distress Syndrome Neutrophils Have a Distinct Phenotype and Are Resistant to Phosphoinositide 3-Kinase Inhibition. Am. J. Respir. Crit. Care Med. 2016, 194, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Orr, Y.; Wilson, D.P.; Taylor, J.M.; Bannon, P.G.; Geczy, C.; Davenport, M.P.; Kritharides, L. A kinetic model of bone marrow neutrophil production that characterizes late phenotypic maturation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1707–R1716. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, L.; Hidalgo, A.; A-Gonzalez, N. Emerging roles of infiltrating granulocytes and monocytes in homeostasis. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [Green Version]
- Geering, B.; Stoeckle, C.; Conus, S.; Simon, H.U. Living and dying for inflammation: Neutrophils, eosinophils, basophils. Trends Immunol. 2013, 34, 398–409. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Worthen, G.S.; Schwab, B., III; Elson, E.L.; Downey, G.P. Mechanics of stimulated neutrophils: Cell stiffening induces retention in capillaries. Science 1989, 245, 183–186. [Google Scholar] [CrossRef]
- Downey, G.P.; Worthen, G.S.; Henson, P.M.; Hyde, D.M. Neutrophil sequestration and migration in localized pulmonary inflammation. Capillary localization and migration across the interalveolar septum. Am. Rev. Respir. Dis. 1993, 147, 168–176. [Google Scholar] [CrossRef]
- Hoffmann, K.; Sperling, K.; Olins, A.L.; Olins, D.E. The granulocyte nucleus and lamin B receptor: Avoiding the ovoid. Chromosoma 2007, 116, 227–235. [Google Scholar] [CrossRef]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Olins, A.L.; Herrmann, H.; Lichter, P.; Olins, D.E. Retinoic acid differentiation of HL-60 cells promotes cytoskeletal polarization. Exp. Cell Res. 2000, 254, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins A and C but not lamin B1 regulate nuclear mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lammerding, J. Mechanics of the nucleus. Compr. Physiol. 2011, 1, 783–807. [Google Scholar] [PubMed] [Green Version]
- Rowat, A.C.; Jaalouk, D.E.; Zwerger, M.; Ung, W.L.; Eydelnant, I.A.; Olins, D.E.; Olins, A.L.; Herrmann, H.; Weitz, D.A.; Lammerding, J. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J. Biol. Chem. 2013, 288, 8610–8618. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl. Acad. Sci. USA 2020, 117, 7326–7337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahoz-Beneytez, J.; Elemans, M.; Zhang, Y.; Ahmed, R.; Salam, A.; Block, M.; Niederalt, C.; Asquith, B.; Macallan, D. Human neutrophil kinetics: Modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 2016, 127, 3431–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broers, J.L.; Peeters, E.A.; Kuijpers, H.J.; Endert, J.; Bouten, C.V.; Oomens, C.W.; Baaijens, F.P.; Ramaekers, F.C. Decreased mechanical stiffness in LMNA−/− cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum. Mol. Genet. 2004, 13, 2567–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Shah, P.; Wolf, K.; Lammerding, J. Bursting the Bubble—Nuclear Envelope Rupture as a Path to Genomic Instability? Trends Cell Biol. 2017, 27, 546–555. [Google Scholar] [CrossRef]
- Majewski, P.; Majchrzak-Gorecka, M.; Grygier, B.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Cichy, J. Inhibitors of Serine Proteases in Regulating the Production and Function of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 261. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Farley, K.; Stolley, J.M.; Zhao, P.; Cooley, J.; Remold-O’Donnell, E. A serpinB1 regulatory mechanism is essential for restricting neutrophil extracellular trap generation. J. Immunol. 2012, 189, 4574–4581. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Chojnowski, A.; Boudier, T.; Lim, J.S.; Ahmed, S.; Ser, Z.; Stewart, C.; Burke, B. A-type Lamins Form Distinct Filamentous Networks with Differential Nuclear Pore Complex Associations. Curr. Biol. 2016, 26, 2651–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.R.; Gordon, S. Monocyte heterogeneity and innate immunity. Immunity 2003, 19, 2–4. [Google Scholar] [CrossRef] [Green Version]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Mass, E. Delineating the origins, developmental programs and homeostatic functions of tissue-resident macrophages. Int. Immunol. 2018, 30, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasrollahzadeh, E.; Razi, S.; Keshavarz-Fathi, M.; Mazzone, M.; Rezaei, N. Pro-tumorigenic functions of macrophages at the primary, invasive and metastatic tumor site. Cancer Immunol. Immunother. 2020, 69, 1673–1697. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef]
- Verreck, F.A.; de Boer, T.; Langenberg, D.M.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M. The many faces of macrophage activation. J. Leukoc. Biol. 2003, 73, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Sobenin, I.A.; Orekhov, A.N. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int. J. Cardiol. 2015, 184, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.F.; Gerber, J.S.; Mosser, D.M. Modulating macrophage function with IgG immune complexes. J. Endotoxin. Res. 2002, 8, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.L.; Singer, K.; Lumeng, C.N. Adipose tissue macrophages: Phenotypic plasticity and diversity in lean and obese states. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.C.; Kim, M.S.; Pae, M.; Yamamoto, Y.; Eberle, D.; Shimada, T.; Kamei, N.; Park, H.S.; Sasorith, S.; Woo, J.R.; et al. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab. 2016, 23, 685–698. [Google Scholar] [CrossRef]
- Chiang, S.H.; Bazuine, M.; Lumeng, C.N.; Geletka, L.M.; Mowers, J.; White, N.M.; Ma, J.T.; Zhou, J.; Qi, N.; Westcott, D.; et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009, 138, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Bayona, P.W.; Kim, M.; Chang, J.; Hong, S.; Park, Y.; Budiman, A.; Kim, Y.J.; Choi, C.Y.; Kim, W.S.; et al. Macrophage Lamin A/C Regulates Inflammation and the Development of Obesity-Induced Insulin Resistance. Front. Immunol. 2018, 9, 696. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Alvey, C.M.; Spinler, K.R.; Irianto, J.; Pfeifer, C.R.; Hayes, B.; Xia, Y.; Cho, S.; Dingal, P.; Hsu, J.; Smith, L.; et al. SIRPA-Inhibited, Marrow-Derived Macrophages Engorge, Accumulate, and Differentiate in Antibody-Targeted Regression of Solid Tumors. Curr. Biol. 2017, 27, 2065–2077.e6. [Google Scholar] [CrossRef] [Green Version]
- Andrechak, J.C.; Dooling, L.J.; Discher, D.E. The macrophage checkpoint CD47: SIRPalpha for recognition of ‘self’ cells: From clinical trials of blocking antibodies to mechanobiological fundamentals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180217. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Edwards, R.; Tucci, S.; Bu, P.; Milsom, J.; Lee, S.; Edelmann, W.; Gumus, Z.H.; Shen, X.; Lipkin, S. Chemokine 25-induced signaling suppresses colon cancer invasion and metastasis. J. Clin. Investig. 2012, 122, 3184–3196. [Google Scholar] [CrossRef] [PubMed]
- Imperial, R.; Ahmed, Z.; Toor, O.M.; Erdogan, C.; Khaliq, A.; Case, P.; Case, J.; Kennedy, K.; Cummings, L.S.; Melton, N.; et al. Comparative proteogenomic analysis of right-sided colon cancer, left-sided colon cancer and rectal cancer reveals distinct mutational profiles. Mol. Cancer 2018, 17, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, V.L.; Jeraldo, P.; Chen, J.; Mundy, M.; Yao, J.; Priya, S.; Keeney, G.; Lyke, K.; Ridlon, J.; White, B.A.; et al. Distinct microbes, metabolites, and ecologies define the microbiome in deficient and proficient mismatch repair colorectal cancers. Genome Med. 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yuan, Y.; Yang, F.; Wang, Y.; Zhu, X.; Wang, Z.; Zheng, S.; Wan, D.; He, J.; Wang, J.; et al. Expert consensus on multidisciplinary therapy of colorectal cancer with lung metastases (2019 edition). J. Hematol. Oncol. 2019, 12, 16. [Google Scholar] [CrossRef]
- Bu, P.; Chen, K.Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [Google Scholar] [CrossRef] [Green Version]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Lin, X.J.; Yang, X.J.; Zhou, L.; He, S.; Zhuang, S.M.; Yang, J. A novel AP-1/miR-101 regulatory feedback loop and its implication in the migration and invasion of hepatoma cells. Nucleic Acids Res. 2014, 42, 12041–12051. [Google Scholar] [CrossRef] [Green Version]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Ivorra, C.; Kubicek, M.; Gonzalez, J.M.; Sanz-Gonzalez, S.M.; Alvarez-Barrientos, A.; O’Connor, J.E.; Burke, B.; Andres, V. A mechanism of AP-1 suppression through interaction of c-Fos with lamin A/C. Genes Dev. 2006, 20, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, J.M.; Navarro-Puche, A.; Casar, B.; Crespo, P.; Andres, V. Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J. Cell Biol. 2008, 183, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ma, Y.M.; Zheng, P.S.; Zhang, P. GDF15 promotes the proliferation of cervical cancer cells by phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J. Exp. Clin. Cancer Res. 2018, 37, 80. [Google Scholar] [CrossRef] [PubMed]
- Corre, J.; Labat, E.; Espagnolle, N.; Hebraud, B.; Avet-Loiseau, H.; Roussel, M.; Huynh, A.; Gadelorge, M.; Cordelier, P.; Klein, B.; et al. Bioactivity and prognostic significance of growth differentiation factor GDF15 secreted by bone marrow mesenchymal stem cells in multiple myeloma. Cancer Res. 2012, 72, 1395–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.S.; Song, M.; Bezawada, N.; Wu, K.; Garcia-Albeniz, X.; Morikawa, T.; Fuchs, C.S.; Ogino, S.; Giovannucci, E.L.; Chan, A.T. A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J. Natl. Cancer Inst. 2014, 106, dju016. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Hao, K.; Li, Z.; Ma, R.; Zhou, Y.; Zhou, Z.; Wei, M.; Liao, Y.; Dai, Y.; Yang, Y.; et al. c-Fos separation from Lamin A/C by GDF15 promotes colon cancer invasion and metastasis in inflammatory microenvironment. J. Cell. Physiol. 2020, 235, 4407–4421. [Google Scholar] [CrossRef]
- Serafim, T.D.; Coutinho-Abreu, I.V.; Oliveira, F.; Meneses, C.; Kamhawi, S.; Valenzuela, J.G. Sequential blood meals promote Leishmania replication and reverse metacyclogenesis augmenting vector infectivity. Nat. Microbiol. 2018, 3, 548–555. [Google Scholar] [CrossRef]
- Liu, D.; Uzonna, J.E. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front. Cell. Infect. Microbiol. 2012, 2, 83. [Google Scholar] [CrossRef] [Green Version]
- Tomiotto-Pellissier, F.; Bortoleti, B.; Assolini, J.P.; Goncalves, M.D.; Carloto, A.C.M.; Miranda-Sapla, M.M.; Conchon-Costa, I.; Bordignon, J.; Pavanelli, W.R. Macrophage Polarization in Leishmaniasis: Broadening Horizons. Front. Immunol. 2018, 9, 2529. [Google Scholar] [CrossRef] [Green Version]
- Loria-Cervera, E.N.; Andrade-Narvaez, F. The role of monocytes/macrophages in Leishmania infection: A glance at the human response. Acta Trop. 2020, 207, 105456. [Google Scholar] [CrossRef]
- Ovalle-Bracho, C.; Londono-Barbosa, D.A.; Franco-Munoz, C.; Clavijo-Ramirez, C. Functional evaluation of gene silencing on macrophages derived from U937 cells using interference RNA (shRNA) in a model of macrophages infected with Leishmania (Viannia) braziliensis. Parasitology 2015, 142, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Odgren, P.R.; Pratt, C.H.; Mackay, C.A.; Mason-Savas, A.; Curtain, M.; Shopland, L.; Ichicki, T.; Sundberg, J.P.; Donahue, L.R. Disheveled hair and ear (Dhe), a spontaneous mouse Lmna mutation modeling human laminopathies. PLoS ONE 2010, 5, e9959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, L.; Jiang, T.T.; Kinder, J.M.; Ertelt, J.M.; Way, S.S. Infection susceptibility and immune senescence with advancing age replicated in accelerated aging Lmna(Dhe) mice. Aging Cell 2015, 14, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, H.; Xu, M.; Han, F.; Tian, C.; Kim, S.; Fredman, E.; Zhang, J.; Benedict-Alderfer, C.; Zheng, Q.Y. Pathological features in the LmnaDhe/+ mutant mouse provide a novel model of human otitis media and laminopathies. Am. J. Pathol. 2012, 181, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Emerman, M. Retroviral infection of non-dividing cells: Old and new perspectives. Virology 2006, 344, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Eckstein, D.A.; Penn, M.L.; Korin, Y.D.; Scripture-Adams, D.D.; Zack, J.A.; Kreisberg, J.F.; Roederer, M.; Sherman, M.P.; Chin, P.S.; Goldsmith, M.A. HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 2001, 15, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Balliet, J.W.; Kolson, D.L.; Eiger, G.; Kim, F.M.; McGann, K.A.; Srinivasan, A.; Collman, R. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: Mutational analysis of a primary HIV-1 isolate. Virology 1994, 200, 623–631. [Google Scholar] [CrossRef]
- de Noronha, C.M.; Sherman, M.P.; Lin, H.W.; Cavrois, M.V.; Moir, R.D.; Goldman, R.D.; Greene, W.C. Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 2001, 294, 1105–1108. [Google Scholar] [CrossRef]
- Sun, W.W.; Jiao, S.; Sun, L.; Zhou, Z.; Jin, X.; Wang, J.H. SUN2 Modulates HIV-1 Infection and Latency through Association with Lamin A/C To Maintain the Repressive Chromatin. mBio 2018, 9, e02408-17. [Google Scholar] [CrossRef] [Green Version]
- Donahue, D.A.; Amraoui, S.; di Nunzio, F.; Kieffer, C.; Porrot, F.; Opp, S.; Diaz-Griffero, F.; Casartelli, N.; Schwartz, O. SUN2 Overexpression Deforms Nuclear Shape and Inhibits HIV. J. Virol. 2016, 90, 4199–4214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurotaki, D.; Yoshida, H.; Tamura, T. Epigenetic and transcriptional regulation of osteoclast differentiation. Bone 2020, 138, 115471. [Google Scholar] [CrossRef]
- Xiong, L.; Zhao, K.; Cao, Y.; Guo, H.H.; Pan, J.X.; Yang, X.; Ren, X.; Mei, L.; Xiong, W.C. Linking skeletal muscle aging with osteoporosis by lamin A/C deficiency. PLoS Biol. 2020, 18, e3000731. [Google Scholar] [CrossRef]
- Gargiuli, C.; Schena, E.; Mattioli, E.; Columbaro, M.; D’Apice, M.R.; Novelli, G.; Greggi, T.; Lattanzi, G. Lamins and bone disorders: Current understanding and perspectives. Oncotarget 2018, 9, 22817–22831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, S.W.; Lovibond, A.C. Current insights into the role of transforming growth factor-beta in bone resorption. Mol. Cell. Endocrinol. 2005, 243, 19–26. [Google Scholar] [CrossRef]
- Yang, T.; Grafe, I.; Bae, Y.; Chen, S.; Chen, Y.; Bertin, T.K.; Jiang, M.M.; Ambrose, C.G.; Lee, B. E-selectin ligand 1 regulates bone remodeling by limiting bioactive TGF-beta in the bone microenvironment. Proc. Natl. Acad. Sci. USA 2013, 110, 7336–7341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallas, S.L.; Park-Snyder, S.; Miyazono, K.; Twardzik, D.; Mundy, G.R.; Bonewald, L.F. Characterization and autoregulation of latent transforming growth factor beta (TGF beta) complexes in osteoblast-like cell lines. Production of a latent complex lacking the latent TGF beta-binding protein. J. Biol. Chem. 1994, 269, 6815–6821. [Google Scholar]
- Hering, S.; Isken, E.; Knabbe, C.; Janott, J.; Jost, C.; Pommer, A.; Muhr, G.; Schatz, H.; Pfeiffer, A.F. TGFbeta1 and TGFbeta2 mRNA and protein expression in human bone samples. Exp. Clin. Endocrinol. Diabetes 2001, 109, 217–226. [Google Scholar] [CrossRef]
- Oreffo, R.O.; Mundy, G.R.; Seyedin, S.M.; Bonewald, L.F. Activation of the bone-derived latent TGF beta complex by isolated osteoclasts. Biochem. Biophys. Res. Commun. 1989, 158, 817–823. [Google Scholar] [CrossRef]
- Zini, N.; Avnet, S.; Ghisu, S.; Maraldi, N.M.; Squarzoni, S.; Baldini, N.; Lattanzi, G. Effects of prelamin A processing inhibitors on the differentiation and activity of human osteoclasts. J. Cell. Biochem. 2008, 105, 34–40. [Google Scholar] [CrossRef]
- Evangelisti, C.; Bernasconi, P.; Cavalcante, P.; Cappelletti, C.; D’Apice, M.R.; Sbraccia, P.; Novelli, G.; Prencipe, S.; Lemma, S.; Baldini, N.; et al. Modulation of TGFbeta 2 levels by lamin A in U2-OS osteoblast-like cells: Understanding the osteolytic process triggered by altered lamins. Oncotarget 2015, 6, 7424–7437. [Google Scholar] [CrossRef] [Green Version]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutertre, C.A.; Wang, L.F.; Ginhoux, F. Aligning bona fide dendritic cell populations across species. Cell Immunol. 2014, 291, 3–10. [Google Scholar] [CrossRef]
- Zernecke, A. Dendritic cells in atherosclerosis: Evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Pulido, J.; Zernecke, A. Antigen-presenting dendritic cells in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 25–31. [Google Scholar] [CrossRef]
- Greter, M.; Helft, J.; Chow, A.; Hashimoto, D.; Mortha, A.; Agudo-Cantero, J.; Bogunovic, M.; Gautier, E.L.; Miller, J.; Leboeuf, M.; et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 2012, 36, 1031–1046. [Google Scholar] [CrossRef] [Green Version]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic cells: A link between innate and adaptive immunity. J. Clin. Immunol. 1999, 19, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Linking innate and adaptive immunity. Nat. Med. 1999, 5, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Bousso, P. T-cell activation by dendritic cells in the lymph node: Lessons from the movies. Nat. Rev. Immunol. 2008, 8, 675–684. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions about dendritic cells: Past, present, and future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldo, C.R., Jr.; Piazza, P. Virus infection of dendritic cells: Portal for host invasion and host defense. Trends Microbiol. 2004, 12, 337–345. [Google Scholar] [CrossRef]
- Pollara, G.; Kwan, A.; Newton, P.J.; Handley, M.E.; Chain, B.M.; Katz, D.R. Dendritic cells in viral pathogenesis: Protective or defective? Int. J. Exp. Pathol. 2005, 86, 187–204. [Google Scholar] [CrossRef]
- Rechenchoski, D.Z.; Faccin-Galhardi, L.C.; Linhares, R.E.C.; Nozawa, C. Herpesvirus: An underestimated virus. Folia Microbiol. 2017, 62, 151–156. [Google Scholar] [CrossRef]
- Whitley, R.J.; Roizman, B. Herpes simplex virus infections. Lancet 2001, 357, 1513–1518. [Google Scholar] [CrossRef]
- Mou, F.; Wills, E.G.; Park, R.; Baines, J.D. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J. Virol. 2008, 82, 8094–8104. [Google Scholar] [CrossRef] [Green Version]
- Mou, F.; Forest, T.; Baines, J.D. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 2007, 81, 6459–6470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.T.; Tan, H.L.; Huang, Q.; Ong, C.N.; Shen, H.M. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 2009, 5, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.; Baines, J.D. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 2006, 80, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, A.; Grosche, L.; Krawczyk, A.; Muhl-Zurbes, P.; Drassner, C.; Duthorn, A.; Kummer, M.; Hasenberg, M.; Voortmann, S.; Jastrow, H.; et al. Autophagic degradation of lamins facilitates the nuclear egress of herpes simplex virus type 1. J. Cell Biol. 2019, 218, 508–523. [Google Scholar] [CrossRef]
- McFarlane, S.; Aitken, J.; Sutherland, J.S.; Nicholl, M.J.; Preston, V.G.; Preston, C.M. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J. Virol. 2011, 85, 4212–4221. [Google Scholar] [CrossRef] [Green Version]
- Mikloska, Z.; Bosnjak, L.; Cunningham, A.L. Immature monocyte-derived dendritic cells are productively infected with herpes simplex virus type 1. J. Virol. 2001, 75, 5958–5964. [Google Scholar] [CrossRef] [Green Version]
- Kruse, M.; Rosorius, O.; Kratzer, F.; Stelz, G.; Kuhnt, C.; Schuler, G.; Hauber, J.; Steinkasserer, A. Mature dendritic cells infected with herpes simplex virus type 1 exhibit inhibited T-cell stimulatory capacity. J. Virol. 2000, 74, 7127–7136. [Google Scholar] [CrossRef] [Green Version]
- Goldwich, A.; Prechtel, A.T.; Muhl-Zurbes, P.; Pangratz, N.M.; Stossel, H.; Romani, N.; Steinkasserer, A.; Kummer, M. Herpes simplex virus type I (HSV-1) replicates in mature dendritic cells but can only be transferred in a cell-cell contact-dependent manner. J. Leukoc. Biol. 2011, 89, 973–979. [Google Scholar] [CrossRef]
- Pous, C.; Codogno, P. Lysosome positioning coordinates mTORC1 activity and autophagy. Nat. Cell Biol. 2011, 13, 342–344. [Google Scholar] [CrossRef]
- Niedergang, F. Dendritic cells mature to resist lamin degradation and herpes virus release. J. Cell Biol. 2019, 218, 387–388. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Wolf, K. Cancer cell migration in 3D tissue: Negotiating space by proteolysis and nuclear deformability. Cell Adh. Migr. 2015, 9, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Pflicke, H.; Sixt, M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J. Exp. Med. 2009, 206, 2925–2935. [Google Scholar] [CrossRef] [Green Version]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketelhuth, D.F.; Hansson, G.K. Adaptive Response of T and B Cells in Atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos-Moran, E.; Blas-Rus, N.; Martin-Cofreces, N.B.; Sanchez-Madrid, F. Orchestrating Lymphocyte Polarity in Cognate Immune Cell-Cell Interactions. Int. Rev. Cell Mol. Biol. 2016, 327, 195–261. [Google Scholar] [PubMed] [Green Version]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and memory T-cell differentiation: Implications for vaccine development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.; Sarkar, S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2-A Balancing Act. Front. Immunol. 2018, 9, 2987. [Google Scholar] [CrossRef] [Green Version]
- van Panhuys, N.; Klauschen, F.; Germain, R.N. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity 2014, 41, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Toribio-Fernandez, R.; Zorita, V.; Herrero-Fernandez, B.; Gonzalez-Granado, J.M. An In Vivo Mouse Model to Measure Naive CD4 T Cell Activation, Proliferation and Th1 Differentiation Induced by Bone Marrow-derived Dendritic Cells. J. Vis. Exp. 2018, 58118. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Paul, W.E. CD4 T cells: Fates, functions, and faults. Blood 2008, 112, 1557–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Curado, S.; Mayya, V.; Dustin, M.L. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim. Biophys. Acta 2014, 1838, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Martin-Cofreces, N.B.; Baixauli, F.; Sanchez-Madrid, F. Immune synapse: Conductor of orchestrated organelle movement. Trends Cell Biol. 2014, 24, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, J.S.; Frock, R.L.; Mamman, S.A.; Fink, P.J.; Kennedy, B.K. Cell-extrinsic defective lymphocyte development in Lmna(−/−) mice. PLoS ONE 2010, 5, e10127. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Egawa, G.; Grabbe, S.; Kabashima, K. Update of immune events in the murine contact hypersensitivity model: Toward the understanding of allergic contact dermatitis. J. Investig. Dermatol. 2013, 133, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, J.; Calvo, F.; Gonzalez, J.M.; Casar, B.; Andres, V.; Crespo, P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 2010, 191, 967–979. [Google Scholar] [CrossRef] [Green Version]
- Broers, J.L.; Ramaekers, F.C. The role of the nuclear lamina in cancer and apoptosis. Adv. Exp. Med. Biol. 2014, 773, 27–48. [Google Scholar]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997, 89, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Toribio-Fernandez, R.; Zorita, V.; Rocha-Perugini, V.; Iborra, S.; Martinez Del Hoyo, G.; Chevre, R.; Dorado, B.; Sancho, D.; Sanchez-Madrid, F.; Andres, V.; et al. Lamin A/C augments Th1 differentiation and response against vaccinia virus and Leishmania major. Cell Death Dis. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Panhuys, N. TCR Signal Strength Alters T-DC Activation and Interaction Times and Directs the Outcome of Differentiation. Front. Immunol. 2016, 7, 6. [Google Scholar] [CrossRef]
- Kouznetsova, V.L.; Tchekanov, A.; Li, X.; Yan, X.; Tsigelny, I.F. Polycomb repressive 2 complex-Molecular mechanisms of function. Protein. Sci. 2019, 28, 1387–1399. [Google Scholar] [CrossRef]
- van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The Complexity of PRC2 Subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morinobu, A.; Kanno, Y.; O’Shea, J.J. Discrete roles for histone acetylation in human T helper 1 cell-specific gene expression. J. Biol. Chem. 2004, 279, 40640–40646. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.; Collas, P. Nuclear lamins: Making contacts with promoters. Nucleus 2013, 4, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Ronningen, T.; Shah, A.; Oldenburg, A.R.; Vekterud, K.; Delbarre, E.; Moskaug, J.O.; Collas, P. Prepatterning of differentiation-driven nuclear lamin A/C-associated chromatin domains by GlcNAcylated histone H2B. Genome Res. 2015, 25, 1825–1835. [Google Scholar] [CrossRef] [Green Version]
- Tse, K.; Tse, H.; Sidney, J.; Sette, A.; Ley, K. T cells in atherosclerosis. Int. Immunol. 2013, 25, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Khare, A.; Krishnamoorthy, N.; Qi, Z.; Ray, P. Regulatory T cells in many flavors control asthma. Mucosal Immunol. 2010, 3, 216–229. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, M.; Thorp, E.; Hansson, G.K.; Tabas, I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J. Clin. Investig. 2013, 123, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.M.; Jack, R.S.; Wunderlich, F.T.; Bruning, J.C.; Muller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, H.Z.; Sasaki, N.; Hirata, K.I. Regulatory T Cell Immunity in Atherosclerosis. Acta Med. Indones 2017, 49, 63–68. [Google Scholar]
- Arce-Sillas, A.; Alvarez-Luquin, D.D.; Tamaya-Dominguez, B.; Gomez-Fuentes, S.; Trejo-Garcia, A.; Melo-Salas, M.; Cardenas, G.; Rodriguez-Ramirez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1720827. [Google Scholar] [CrossRef] [Green Version]
- Tselios, K.; Sarantopoulos, A.; Gkougkourelas, I.; Boura, P. T regulatory cells: A promising new target in atherosclerosis. Crit. Rev. Immunol. 2014, 34, 389–397. [Google Scholar]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Gondek, D.C.; Lu, L.F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Garin, M.I.; Chu, C.C.; Golshayan, D.; Cernuda-Morollon, E.; Wait, R.; Lechler, R.I. Galectin-1: A key effector of regulation mediated by CD4+CD25+ T cells. Blood 2007, 109, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Ye, F.; Jiang, Z.; Chu, Y.; Xiong, S.; Wang, Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ. 2007, 14, 2076–2084. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F.; Finotto, S.; Glimcher, L.H. The role of Th1/Th2 polarization in mucosal immunity. Nat. Med. 2002, 8, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 2006, 212, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Shahar, N.; Larisch, S. Inhibiting the inhibitors: Targeting anti-apoptotic proteins in cancer and therapy resistance. Drug Resist Updat. 2020, 52, 100712. [Google Scholar] [CrossRef]
- Lindenboim, L.; Zohar, H.; Worman, H.J.; Stein, R. The nuclear envelope: Target and mediator of the apoptotic process. Cell Death Discov. 2020, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Lindenboim, L.; Sasson, T.; Worman, H.J.; Borner, C.; Stein, R. Cellular stress induces Bax-regulated nuclear bubble budding and rupture followed by nuclear protein release. Nucleus 2014, 5, 527–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzidi, S.; Brech, A.; Sioud, M.; Li, X.; Suo, Z.; Nesland, J.M.; Peng, Q. Lamin A/C cleavage by caspase-6 activation is crucial for apoptotic induction by photodynamic therapy with hexaminolevulinate in human B-cell lymphoma cells. Cancer Lett. 2013, 339, 25–32. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Rius, C.; Gonzalez-Granado, J.M. Lamin A/C and the Immune System: One Intermediate Filament, Many Faces. Int. J. Mol. Sci. 2020, 21, 6109. https://doi.org/10.3390/ijms21176109
Saez A, Herrero-Fernandez B, Gomez-Bris R, Somovilla-Crespo B, Rius C, Gonzalez-Granado JM. Lamin A/C and the Immune System: One Intermediate Filament, Many Faces. International Journal of Molecular Sciences. 2020; 21(17):6109. https://doi.org/10.3390/ijms21176109
Chicago/Turabian StyleSaez, Angela, Beatriz Herrero-Fernandez, Raquel Gomez-Bris, Beatriz Somovilla-Crespo, Cristina Rius, and Jose M. Gonzalez-Granado. 2020. "Lamin A/C and the Immune System: One Intermediate Filament, Many Faces" International Journal of Molecular Sciences 21, no. 17: 6109. https://doi.org/10.3390/ijms21176109
APA StyleSaez, A., Herrero-Fernandez, B., Gomez-Bris, R., Somovilla-Crespo, B., Rius, C., & Gonzalez-Granado, J. M. (2020). Lamin A/C and the Immune System: One Intermediate Filament, Many Faces. International Journal of Molecular Sciences, 21(17), 6109. https://doi.org/10.3390/ijms21176109