ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease


Abstract
:1. Scope of This Review
2. Introduction
3. Structures and Functions of apoE Isoforms
4. CNS apoE Protein
5. Effects of apoE Isoforms on Cholesterol Synthesis and Transport/Efflux
6. Effects of apoE Isoforms on Lipid Homeostasis
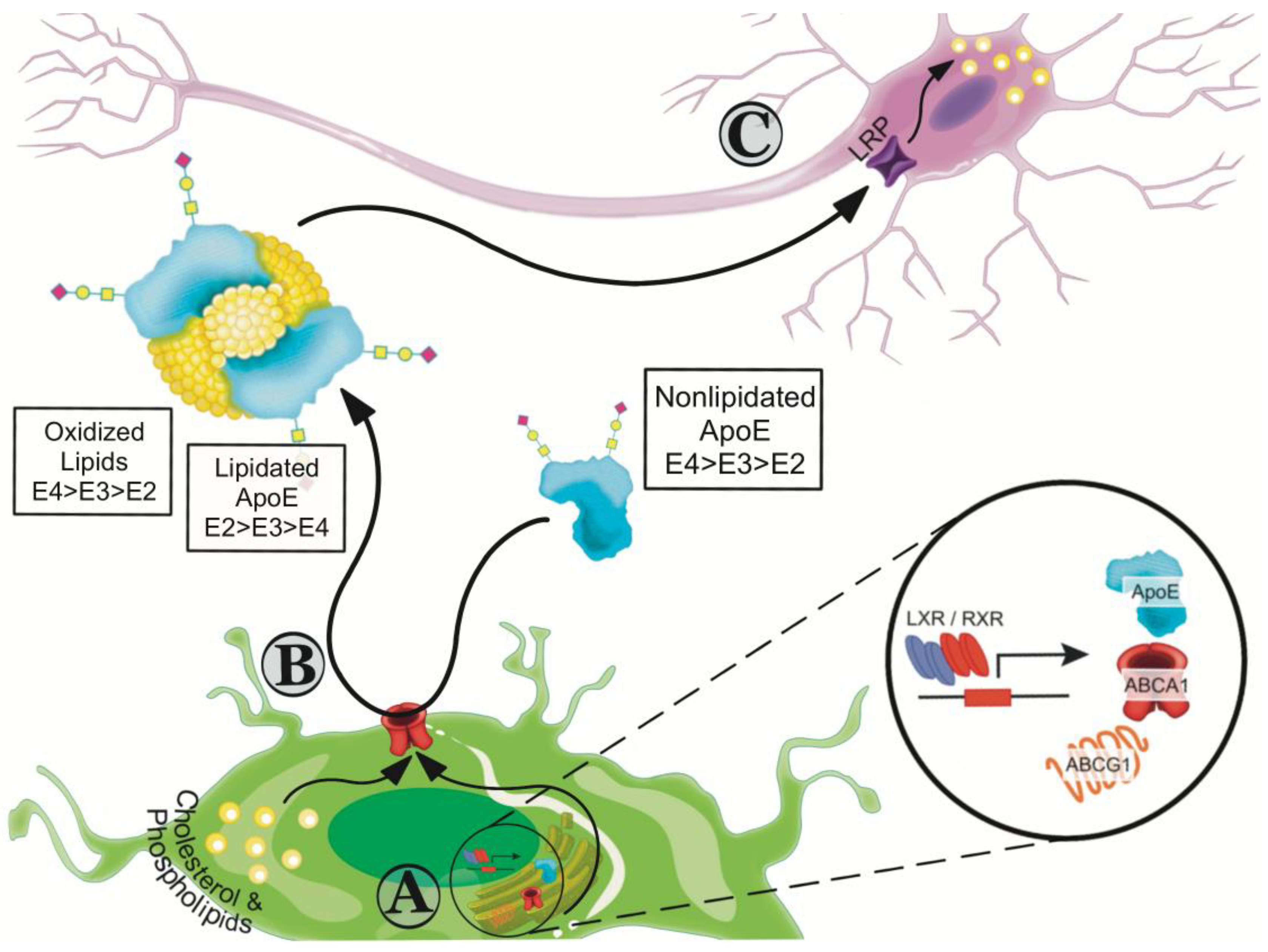
7. Effects of apoE Isoforms on Lipidation
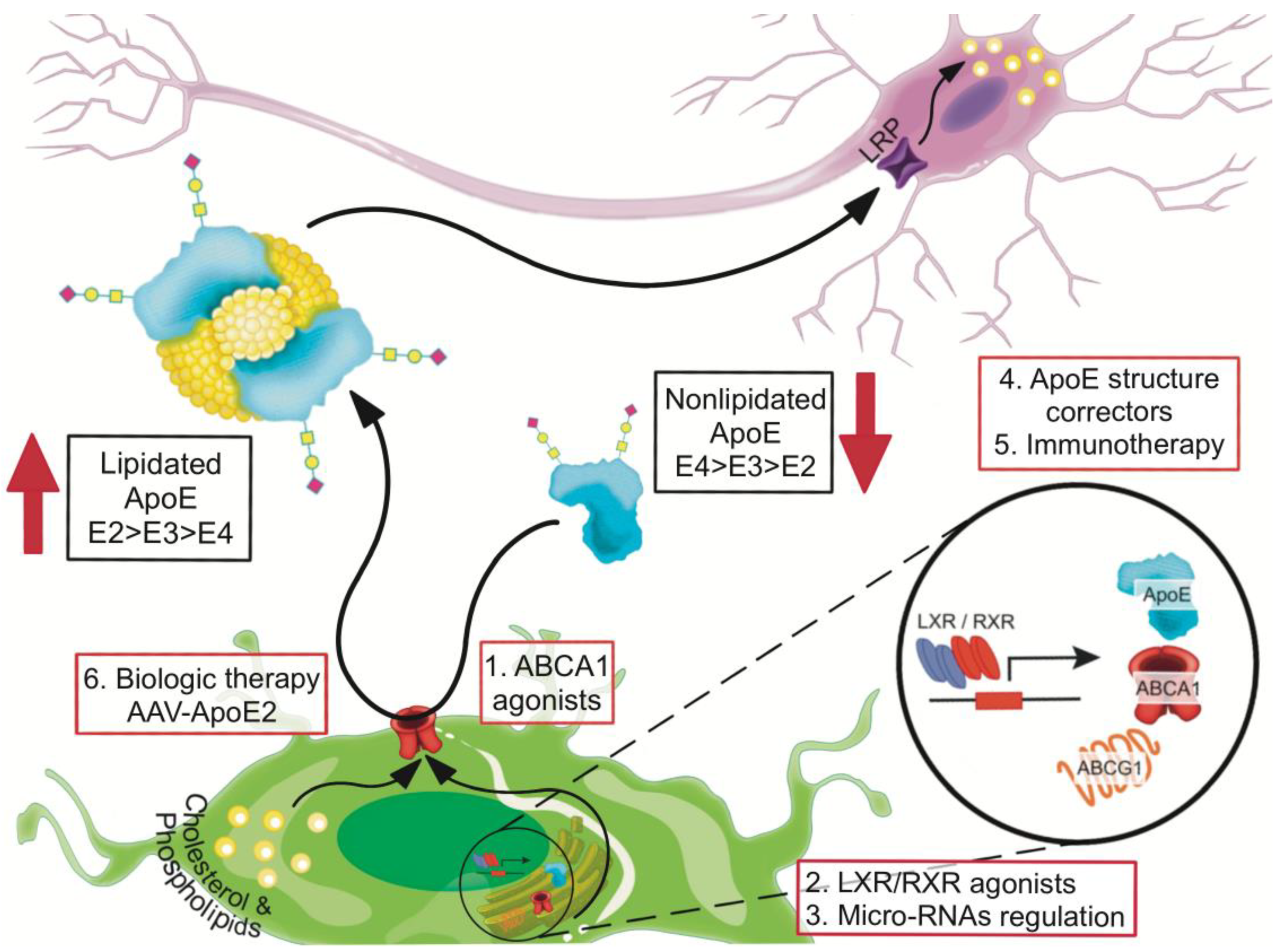
8. Recalibrating apoE Functions by Increasing Lipidation
8.1. Small Molecules that Enhance ABCA1-Mediated apoE4 Lipidation
8.2. Liver X Receptor (LXR) and Retinoid X Receptor (RXR) Agonists
8.3. Small Molecules as apoE4 Structure Correctors
8.4. Anti-apoE4 Immunotherapy
8.5. Recalibrating apoE Function by Using AAV-APOE2 Biologic Therapy
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ABCA1 | ATP-binding cassette transporter A1 |
| ABCG1 | ATP-binding cassette transporter G1 |
| apoE | Apolipoprotein E protein |
| APOE | Apolipoprotein E gene |
| APP | Amyloid precursor protein |
| ARF6 | ADP-ribosylation factor 6 |
| Aβ | Amyloid β |
| BBB | Blood-brain barrier |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CYP46A1 | Cholesterol 24S-hydroxylase |
| FRET | Fluorescence resonance energy transfer |
| HD | Huntington’s disease |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| LRP | LDL receptor-related protein |
| LXR | Liver X receptors |
| NCP | Niemann-Pick type C |
| PD | Parkinson’s disease |
| PPAR-γ | Peroxisome proliferator-activated receptor-γ |
| ROS | Reactive Oxygen Species |
| RXR | Retinoid X receptor |
| SLOS | Smith-Lemli Opitz syndrome |
| SREBP-1 | Sterol regulatory element-binding proteins |
| VLDL | Very-low-density lipoprotein |
References
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, A.L.; Chidgey, C.; Peng, P.; Masters, C.L.; Roberts, B.R. The Neurobiology and Age-Related Prevalence of the epsilon4 Allele of Apolipoprotein E in Alzheimer’s Disease Cohorts. J. Mol. Neurosci. 2016, 60, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Tensaouti, Y.; Yu, T.S.; Kernie, S.G. Apolipoprotein E regulates the maturation of injury-induced adult-born hippocampal neurons following traumatic brain injury. PLoS ONE 2020, 15, e0229240. [Google Scholar] [CrossRef] [Green Version]
- McFadyen, C.A.; Zeiler, F.A.; Newcombe, V.; Synnot, A.; Steyerberg, E.; Gruen, R.L.; Rosand, J.; Palotie, A.; Maas, A.I.R.; Menon, D.K. Apolipoprotein E4 Polymorphism and Outcomes from Traumatic Brain Injury: A Living Systematic Review and Meta-Analysis. J. Neurotrauma 2019. [Google Scholar] [CrossRef] [Green Version]
- Agosta, F.; Vossel, K.A.; Miller, B.L.; Migliaccio, R.; Bonasera, S.J.; Filippi, M.; Boxer, A.L.; Karydas, A.; Possin, K.L.; Gorno-Tempini, M.L. Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2009, 106, 2018–2022. [Google Scholar] [CrossRef] [Green Version]
- Borroni, B.; Perani, D.; Archetti, S.; Agosti, C.; Paghera, B.; Bellelli, G.; Di Luca, M.; Padovani, A. Functional correlates of Apolipoprotein E genotype in Frontotemporal Lobar Degeneration. BMC Neurol. 2006, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Raha-Chowdhury, R.; Henderson, J.W.; Raha, A.A.; Vuono, R.; Bickerton, A.; Jones, E.; Fincham, R.; Allinson, K.; Holland, A.; Zaman, S.H. Choroid Plexus Acts as Gatekeeper for TREM2, Abnormal Accumulation of ApoE, and Fibrillary Tau in Alzheimer’s Disease and in Down Syndrome Dementia. J. Alzheimers Dis. 2019, 69, 91–109. [Google Scholar] [CrossRef] [Green Version]
- Prokopenko, I.; Miyakawa, G.; Zheng, B.; Heikkinen, J.; Petrova Quayle, D.; Udeh-Momoh, C.; Claringbould, A.; Neumann, J.; Haytural, H.; Kaakinen, M.A.; et al. Alzheimer’s disease pathology explains association between dementia with Lewy bodies and APOE-epsilon4/TOMM40 long poly-T repeat allele variants. Alzheimers Dement. 2019, 5, 814–824. [Google Scholar]
- Mahley, R.W. Apolipoprotein E: Remarkable Protein Sheds Light on Cardiovascular and Neurological Diseases. Clin. Chem. 2017, 63, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef] [PubMed]
- Zannis, V.I.; Breslow, J.L.; Utermann, G.; Mahley, R.W.; Weisgraber, K.H.; Havel, R.J.; Goldstein, J.L.; Brown, M.S.; Schonfeld, G.; Hazzard, W.R.; et al. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J. Lipid Res. 1982, 23, 911–914. [Google Scholar]
- Chen, J.; Li, Q.; Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA 2011, 108, 14813–14818. [Google Scholar] [CrossRef] [Green Version]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Henry, N.; Krammer, E.M.; Stengel, F.; Adams, Q.; Van Liefferinge, F.; Hubin, E.; Chaves, R.; Efremov, R.; Aebersold, R.; Vandenbussche, G.; et al. Lipidated apolipoprotein E4 structure and its receptor binding mechanism determined by a combined cross-linking coupled to mass spectrometry and molecular dynamics approach. PLoS Comput. Biol. 2018, 14, e1006165. [Google Scholar] [CrossRef]
- Raussens, V.; Drury, J.; Forte, T.M.; Choy, N.; Goormaghtigh, E.; Ruysschaert, J.M.; Narayanaswami, V. Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem. J. 2005, 387, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Narayanaswami, V.; Maiorano, J.N.; Dhanasekaran, P.; Ryan, R.O.; Phillips, M.C.; Lund-Katz, S.; Davidson, W.S. Helix orientation of the functional domains in apolipoprotein e in discoidal high density lipoprotein particles. J. Biol. Chem. 2004, 279, 14273–14279. [Google Scholar] [CrossRef] [Green Version]
- Saito, H.; Dhanasekaran, P.; Baldwin, F.; Weisgraber, K.H.; Lund-Katz, S.; Phillips, M.C. Lipid binding-induced conformational change in human apolipoprotein E. Evidence for two lipid-bound states on spherical particles. J. Biol. Chem. 2001, 276, 40949–40954. [Google Scholar] [CrossRef] [Green Version]
- Narayanaswami, V.; Ryan, R.O. Molecular basis of exchangeable apolipoprotein function. Biochim. Biophys. Acta 2000, 1483, 15–36. [Google Scholar] [CrossRef]
- Frieden, C.; Wang, H.; Ho, C.M.W. A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc. Natl. Acad. Sci. USA 2017, 114, 6292–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.; Wardell, M.R.; Weisgraber, K.H.; Mahley, R.W.; Agard, D.A. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 1991, 252, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Mau, T.; Weisgraber, K.H.; Wardell, M.R.; Mahley, R.W.; Agard, D.A. Salt bridge relay triggers defective LDL receptor binding by a mutant apolipoprotein. Structure 1994, 2, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Polazzi, E.; Mengoni, I.; Pena-Altamira, E.; Massenzio, F.; Virgili, M.; Petralla, S.; Monti, B. Neuronal Regulation of Neuroprotective Microglial Apolipoprotein E Secretion in Rat In Vitro Models of Brain Pathophysiology. J. Neuropathol. Exp. Neurol. 2015, 74, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Bernardo, A.; Walker, D.; Kanegawa, T.; Mahley, R.W.; Huang, Y. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 2006, 26, 4985–4994. [Google Scholar] [CrossRef]
- Huang, Y.; Weisgraber, K.H.; Mucke, L.; Mahley, R.W. Apolipoprotein E: Diversity of cellular origins, structural and biophysical properties, and effects in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 189–204. [Google Scholar] [CrossRef]
- Metzger, R.E.; LaDu, M.J.; Pan, J.B.; Getz, G.S.; Frail, D.E.; Falduto, M.T. Neurons of the human frontal cortex display apolipoprotein E immunoreactivity: Implications for Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 372–380. [Google Scholar] [CrossRef] [Green Version]
- Boyles, J.K.; Pitas, R.E.; Wilson, E.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Investig. 1985, 76, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Achariyar, T.M.; Li, B.; Peng, W.; Verghese, P.B.; Shi, Y.; McConnell, E.; Benraiss, A.; Kasper, T.; Song, W.; Takano, T.; et al. Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation. Mol. Neurodegener. 2016, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Flowers, S.A.; Grant, O.C.; Woods, R.J.; Rebeck, G.W. O-glycosylation on cerebrospinal fluid and plasma apolipoprotein E differs in the lipid-binding domain. Glycobiology 2019, 30, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.A.; Rebeck, G.W. APOE in the normal brain. Neurobiol. Dis. 2020, 136, 104724. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Meuret, C.; Go, S.; Yassine, H.N.; Nedelkov, D. Simple and Fast Assay for Apolipoprotein E Phenotyping and Glycotyping: Discovering Isoform-Specific Glycosylation in Plasma and Cerebrospinal Fluid. J. Alzheimers Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kockx, M.; Traini, M.; Kritharides, L. Cell-specific production, secretion, and function of apolipoprotein E. J. Mol. Med. 2018, 96, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Pitas, R.E.; Boyles, J.K.; Lee, S.H.; Foss, D.; Mahley, R.W. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta 1987, 917, 148–161. [Google Scholar] [CrossRef]
- Mailly, F.; Davignon, J.; Nestruck, A.C. Analytical isoelectric focusing with immobilized pH gradients of human apolipoprotein E from very low density lipoproteins and total plasma. J. Lipid Res. 1990, 31, 149–155. [Google Scholar]
- DiBattista, A.M.; Dumanis, S.B.; Newman, J.; Rebeck, G.W. Identification and modification of amyloid-independent phenotypes of APOE4 mice. Exp. Neurol. 2016, 280, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Cruchaga, C.; Kauwe, J.S.; Nowotny, P.; Bales, K.; Pickering, E.H.; Mayo, K.; Bertelsen, S.; Hinrichs, A.; Alzheimer’s Disease Neuroimaging Initiative; Fagan, A.M.; et al. Cerebrospinal fluid APOE levels: An endophenotype for genetic studies for Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 4558–4571. [Google Scholar] [CrossRef] [Green Version]
- Riddell, D.R.; Zhou, H.; Atchison, K.; Warwick, H.K.; Atkinson, P.J.; Jefferson, J.; Xu, L.; Aschmies, S.; Kirksey, Y.; Hu, Y.; et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 2008, 28, 11445–11453. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.M.; Han, B.; Liu, F.; Mace, B.E.; Ervin, J.F.; Wu, S.; Koger, D.; Paul, S.; Bales, K.R. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol. Aging 2011, 32, 791–801. [Google Scholar] [CrossRef]
- Martinez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Marchi, C.; Adorni, M.P.; Caffarra, P.; Ronda, N.; Spallazzi, M.; Barocco, F.; Galimberti, D.; Bernini, F.; Zimetti, F. ABCA1-and ABCG1-mediated cholesterol efflux capacity of cerebrospinal fluid is impaired in Alzheimer’s disease. J. Lipid Res. 2019, 60, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Bellosta, S. Cholesterol: Its regulation and role in central nervous system disorders. Cholest. 2012, 2012, 292598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieweg, K.; Schaller, H.; Pfrieger, F.W. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 2009, 109, 125–134. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Moutinho, M.; Landreth, G.E. Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J. Lipid Res. 2017, 58, 1937–1949. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Lin, S.; Beyer, T.P.; Zhang, Y.; Wu, X.; Bales, K.R.; DeMattos, R.B.; May, P.C.; Li, S.D.; Jiang, X.C.; et al. A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein E expression, secretion and cholesterol homeostasis in astrocytes. J. Neurochem. 2004, 88, 623–634. [Google Scholar] [CrossRef]
- Horiuchi, Y.; Ohkawa, R.; Lai, S.J.; Yamazaki, A.; Ikoma, H.; Yano, K.; Kameda, T.; Tozuka, M. Characterization of the cholesterol efflux of apolipoprotein E-containing high-density lipoprotein in THP-1 cells. Biol. Chem. 2019, 400, 209–218. [Google Scholar] [CrossRef]
- Tarr, P.T.; Edwards, P.A. ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP-2. J. Lipid Res. 2008, 49, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, B.L.; Parkinson, P.F.; Racke, M.M.; Hirsch-Reinshagen, V.; Fan, J.; Wong, C.; Stukas, S.; Theroux, L.; Chan, J.Y.; Donkin, J.; et al. ABCG1 influences the brain cholesterol biosynthetic pathway but does not affect amyloid precursor protein or apolipoprotein E metabolism in vivo. J. Lipid Res. 2008, 49, 1254–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, E.G.; Guileyardo, J.M.; Russell, D.W. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc. Natl. Acad. Sci. USA 1999, 96, 7238–7243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. Dysregulation of cholesterol balance in the brain: Contribution to neurodegenerative diseases. Dis. Model. Mech. 2012, 5, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, M.B.; Mukhopadhyay, D. Possible role of apolipoprotein A1 in healing and cell death after neuronal injury. Front. Biosci. (Elite Ed.) 2016, 8, 460–477. [Google Scholar]
- Nathan, B.P.; Jiang, Y.; Wong, G.K.; Shen, F.; Brewer, G.J.; Struble, R.G. Apolipoprotein E4 inhibits, and apolipoprotein E3 promotes neurite outgrowth in cultured adult mouse cortical neurons through the low-density lipoprotein receptor-related protein. Brain Res. 2002, 928, 96–105. [Google Scholar] [CrossRef]
- Pitas, R.E.; Ji, Z.S.; Weisgraber, K.H.; Mahley, R.W. Role of apolipoprotein E in modulating neurite outgrowth: Potential effect of intracellular apolipoprotein E. Biochem. Soc. Trans. 1998, 26, 257–262. [Google Scholar] [CrossRef]
- Zhu, Y.; Bellosta, S.; Langer, C.; Bernini, F.; Pitas, R.E.; Mahley, R.W.; Assmann, G.; von Eckardstein, A. Low-dose expression of a human apolipoprotein E transgene in macrophages restores cholesterol efflux capacity of apolipoprotein E-deficient mouse plasma. Proc. Natl. Acad. Sci. USA 1998, 95, 7585–7590. [Google Scholar] [CrossRef] [Green Version]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lutjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Shinohara, M.; Sato, N. The Roles of Apolipoprotein E, Lipids, and Glucose in the Pathogenesis of Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1128, 85–101. [Google Scholar] [PubMed]
- Roda, A.R.; Montoliu-Gaya, L.; Villegas, S. The Role of Apolipoprotein E Isoforms in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 459–471. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Burchett, J.M.; Restivo, J.L.; Schuler, D.R.; Verghese, P.B.; Mahan, T.E.; Landreth, G.E.; Castellano, J.M.; Jiang, H.; Cirrito, J.R.; et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol. Neurodegener. 2013, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenza, M.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Fenu, S.; Jamot, L.; Strand, A.; Tarditi, A.; Woodman, B.; Racchi, M.; et al. Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J. Neurosci. 2005, 25, 9932–9939. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, H.; Gong, J.S.; Jung, C.G.; Watanabe, A.; Lund-Katz, S.; Phillips, M.C.; Saito, H.; Michikawa, M. Mechanism underlying apolipoprotein E (ApoE) isoform-dependent lipid efflux from neural cells in culture. J. Neurosci. Res. 2009, 87, 2498–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.S.; Morita, S.Y.; Kobayashi, M.; Handa, T.; Fujita, S.C.; Yanagisawa, K.; Michikawa, M. Novel action of apolipoprotein E (ApoE): ApoE isoform specifically inhibits lipid-particle-mediated cholesterol release from neurons. Mol. Neurodegener. 2007, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Matsushima, T.; Satoh, H.; Iso-o, N.; Noto, H.; Togo, M.; Kimura, S.; Hashimoto, Y.; Tsukamoto, K. Isoform-dependent cholesterol efflux from macrophages by apolipoprotein E is modulated by cell surface proteoglycans. Arter. Thromb. Vasc. Biol. 2003, 23, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [Green Version]
- Julia, T.C.W.; Liang, S.A.; Qian, L.; Pipalia, N.H.; Chao, M.J.; Shi, Y.; Bertelsen, S.E.; Kapoor, M.; Marcora, E.; Sikora, E.; et al. Cholesterol and matrisome pathways dysregulated in human APOE ε4 glia. BioRxiv 2019, 99, 713362. [Google Scholar]
- Jeong, W.; Lee, H.; Cho, S.; Seo, J. ApoE4-Induced Cholesterol Dysregulation and Its Brain Cell Type-Specific Implications in the Pathogenesis of Alzheimer’s Disease. Mol. Cells 2019, 42, 739–746. [Google Scholar]
- de Chaves, E.P.; Narayanaswami, V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008, 3, 505–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papassotiropoulos, A.; Lutjohann, D.; Bagli, M.; Locatelli, S.; Jessen, F.; Buschfort, R.; Ptok, U.; Bjorkhem, I.; von Bergmann, K.; Heun, R. 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J. Psychiatr. Res. 2002, 36, 27–32. [Google Scholar] [CrossRef]
- Boehm-Cagan, A.; Bar, R.; Harats, D.; Shaish, A.; Levkovitz, H.; Bielicki, J.K.; Johansson, J.O.; Michaelson, D.M. Differential Effects of apoE4 and Activation of ABCA1 on Brain and Plasma Lipoproteins. PLoS ONE 2016, 11, e0166195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.O.W.; Michaelson, D.M.; Hartmann, T. Omega-3 fatty acids, lipids, and apoE lipidation in Alzheimer’s disease: A rationale for multi-nutrient dementia prevention. J. Lipid Res. 2017, 58, 2083–2101. [Google Scholar] [CrossRef] [Green Version]
- Samieri, C.; Lorrain, S.; Buaud, B.; Vaysse, C.; Berr, C.; Peuchant, E.; Cunnane, S.C.; Barberger-Gateau, P. Relationship between diet and plasma long-chain n-3 PUFAs in older people: Impact of apolipoprotein E genotype. J. Lipid Res. 2013, 54, 2559–2567. [Google Scholar] [CrossRef] [Green Version]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- McDougall, M.; Choi, J.; Magnusson, K.; Truong, L.; Tanguay, R.; Traber, M.G. Chronic vitamin E deficiency impairs cognitive function in adult zebrafish via dysregulation of brain lipids and energy metabolism. Free Radic. Biol. Med. 2017, 112, 308–317. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Johnson, L.A. APOE in Alzheimer’s disease and neurodegeneration. Neurobiol. Dis. 2020, 139, 104847. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Ramassamy, C.; Averill, D.; Beffert, U.; Theroux, L.; Lussier-Cacan, S.; Cohn, J.S.; Christen, Y.; Schoofs, A.; Davignon, J.; Poirier, J. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol. Dis. 2000, 7, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Lauderback, C.M.; Kanski, J.; Hackett, J.M.; Maeda, N.; Kindy, M.S.; Butterfield, D.A. Apolipoprotein E modulates Alzheimer’s Abeta(1-42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res. 2002, 924, 90–97. [Google Scholar] [CrossRef]
- Pedersen, W.A.; Chan, S.L.; Mattson, M.P. A mechanism for the neuroprotective effect of apolipoprotein E: Isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 2000, 74, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Abeta in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Liu, C.C.; Chen, X.F.; Zhang, Y.W.; Xu, H.; Bu, G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol. Neurodegener. 2015, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Hanson, A.J.; Bayer-Carter, J.L.; Green, P.S.; Montine, T.J.; Wilkinson, C.W.; Baker, L.D.; Watson, G.S.; Bonner, L.M.; Callaghan, M.; Leverenz, J.B.; et al. Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides: Randomized clinical trial. JAMA Neurol. 2013, 70, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Heinsinger, N.M.; Gachechiladze, M.A.; Rebeck, G.W. Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J. Neuropathol. Exp. Neurol. 2016, 75, 918–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, C.S.; Atagi, Y.; Yamazaki, Y.; Shinohara, M.; Tachibana, M.; Fu, Y.; Bu, G.; Kanekiyo, T. Apolipoprotein E Inhibits Cerebrovascular Pericyte Mobility through a RhoA Protein-mediated Pathway. J. Biol. Chem. 2015, 290, 14208–14217. [Google Scholar] [CrossRef] [Green Version]
- Koldamova, R.; Fitz, N.F.; Lefterov, I. ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol. Dis. 2014, 72, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Hubin, E.; Verghese, P.B.; van Nuland, N.; Broersen, K. Apolipoprotein E associated with reconstituted high-density lipoprotein-like particles is protected from aggregation. FEBS Lett. 2019, 593, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.Y.; Lin, Y.L.; Huang, Y.C.; Sheu, S.Y.; Lin, T.H.; Tsay, H.J.; Chang, G.G.; Shiao, M.S. Structural variation in human apolipoprotein E3 and E4: Secondary structure, tertiary structure, and size distribution. Biophys. J. 2005, 88, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Perugini, M.A.; Schuck, P.; Howlett, G.J. Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J. Biol. Chem. 2000, 275, 36758–36765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garai, K.; Frieden, C. The association-dissociation behavior of the ApoE proteins: Kinetic and equilibrium studies. Biochemistry 2010, 49, 9533–9541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, M.; Fryer, J.D.; Sullivan, P.M.; Christopher, E.A.; Wahrle, S.E.; DeMattos, R.B.; O’Dell, M.A.; Fagan, A.M.; Lashuel, H.A.; Walz, T.; et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol. Dis. 2005, 19, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Zhao, J.; Atagi, Y.; Nielsen, H.M.; Liu, C.C.; Zheng, H.; Shinohara, M.; Kanekiyo, T.; Bu, G. Apolipoprotein E lipoprotein particles inhibit amyloid-beta uptake through cell surface heparan sulphate proteoglycan. Mol. Neurodegener. 2016, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodart, J.C.; Marr, R.A.; Koistinaho, M.; Gregersen, B.M.; Malkani, S.; Verma, I.M.; Paul, S.M. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Davis, M.D.; Martens, Y.A.; Shinohara, M.; Graff-Radford, N.R.; Younkin, S.G.; Wszolek, Z.K.; Kanekiyo, T.; Bu, G. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017, 26, 2690–2700. [Google Scholar] [CrossRef]
- Peng, D.; Song, C.; Reardon, C.A.; Liao, S.; Getz, G.S. Lipoproteins produced by ApoE-/- astrocytes infected with adenovirus expressing human ApoE. J. Neurochem. 2003, 86, 1391–1402. [Google Scholar] [CrossRef]
- Potier, M.-C.; Hanbouch, L.; Marquer, C. Cholesterol and ApoE in Alzheimer’s disease. Oilseeds Fats Crop. Lipids 2018, 25, 6. [Google Scholar]
- Marquer, C.; Laine, J.; Dauphinot, L.; Hanbouch, L.; Lemercier-Neuillet, C.; Pierrot, N.; Bossers, K.; Le, M.; Corlier, F.; Benstaali, C.; et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol. Neurodegener. 2014, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Burg, V.K.; Grimm, H.S.; Rothhaar, T.L.; Grosgen, S.; Hundsdorfer, B.; Haupenthal, V.J.; Zimmer, V.C.; Mett, J.; Weingartner, O.; Laufs, U.; et al. Plant sterols the better cholesterol in Alzheimer’s disease? A mechanistical study. J. Neurosci. 2013, 33, 16072–16087. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Han, X.; Tanimura, Y.; Hamanaka, H.; Kobayashi, M.; Sakurai, T.; Yokoyama, M.; Wada, K.; Nukina, N.; Fujita, S.C.; et al. Effects of human apolipoprotein E isoforms on the amyloid beta-protein concentration and lipid composition in brain low-density membrane domains. J. Neurochem. 2007, 101, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitz, N.F.; Cronican, A.A.; Lefterov, I.; Koldamova, R. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science 2013, 340, 924-c. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Yoon, H.; Horie, T.; Burchett, J.M.; Restivo, J.L.; Rotllan, N.; Ramirez, C.M.; Verghese, P.B.; Ihara, M.; Hoe, H.S.; et al. microRNA-33 Regulates ApoE Lipidation and Amyloid-beta Metabolism in the Brain. J. Neurosci. 2015, 35, 14717–14726. [Google Scholar] [CrossRef]
- Rawat, V.; Wang, S.; Sima, J.; Bar, R.; Liraz, O.; Gundimeda, U.; Parekh, T.; Chan, J.; Johansson, J.O.; Tang, C.; et al. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J. Neurosci. 2019, 39, 9611–9622. [Google Scholar] [CrossRef]
- Hafiane, A.; Johansson, J.O.; Genest, J. ABCA1 Agonist Mimetic Peptide CS-6253 Induces Microparticles Release From Different Cell Types by ABCA1-Efflux-Dependent Mechanism. Can. J. Cardiol. 2019, 35, 770–781. [Google Scholar] [CrossRef]
- Boehm-Cagan, A.; Bar, R.; Liraz, O.; Bielicki, J.K.; Johansson, J.O.; Michaelson, D.M. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J. Alzheimers Dis. 2016, 54, 1219–1233. [Google Scholar] [CrossRef]
- Handattu, S.P.; Monroe, C.E.; Nayyar, G.; Palgunachari, M.N.; Kadish, I.; van Groen, T.; Anantharamaiah, G.M.; Garber, D.W. In vivo and in vitro effects of an apolipoprotein e mimetic peptide on amyloid-beta pathology. J. Alzheimers Dis. 2013, 36, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Chernick, D.; Ortiz-Valle, S.; Jeong, A.; Swaminathan, S.K.; Kandimalla, K.K.; Rebeck, G.W.; Li, L. High-density lipoprotein mimetic peptide 4F mitigates amyloid-beta-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J. Neurochem. 2018, 147, 647–662. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, R.L.; Movva, R.; Bloedon, L.T.; Duffy, D.; Norris, R.B.; Navab, M.; Fogelman, A.M.; Rader, D.J. Oral Apolipoprotein A-I Mimetic D-4F Lowers HDL-Inflammatory Index in High-Risk Patients: A First-in-Human Multiple-Dose, Randomized Controlled Trial. Clin. Transl. Sci. 2017, 10, 455–469. [Google Scholar] [CrossRef]
- Bloedon, L.T.; Dunbar, R.; Duffy, D.; Pinell-Salles, P.; Norris, R.; DeGroot, B.J.; Movva, R.; Navab, M.; Fogelman, A.M.; Rader, D.J. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J. Lipid Res. 2008, 49, 1344–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Lee, C.Y.; Mandrekar, S.; Wilkinson, B.; Cramer, P.; Zelcer, N.; Mann, K.; Lamb, B.; Willson, T.M.; Collins, J.L.; et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008, 58, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, D.R.; Zhou, H.; Comery, T.A.; Kouranova, E.; Lo, C.F.; Warwick, H.K.; Ring, R.H.; Kirksey, Y.; Aschmies, S.; Xu, J.; et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell Neurosci. 2007, 34, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Koldamova, R.P.; Lefterov, I.M.; Staufenbiel, M.; Wolfe, D.; Huang, S.; Glorioso, J.C.; Walter, M.; Roth, M.G.; Lazo, J.S. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem. 2005, 280, 4079–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona, A.W.; Kodoma, N.; Casali, B.T.; Landreth, G.E. ABCA1 is Necessary for Bexarotene-Mediated Clearance of Soluble Amyloid Beta from the Hippocampus of APP/PS1 Mice. J. Neuroimmune Pharmacol. 2016, 11, 61–72. [Google Scholar] [CrossRef]
- Boehm-Cagan, A.; Michaelson, D.M. Reversal of apoE4-driven brain pathology and behavioral deficits by bexarotene. J. Neurosci. 2014, 34, 7293–7301. [Google Scholar] [CrossRef] [Green Version]
- Tai, L.M.; Koster, K.P.; Luo, J.; Lee, S.H.; Wang, Y.T.; Collins, N.C.; Ben Aissa, M.; Thatcher, G.R.; LaDu, M.J. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 2014, 289, 30538–30555. [Google Scholar] [CrossRef] [Green Version]
- Mariani, M.M.; Malm, T.; Lamb, R.; Jay, T.R.; Neilson, L.; Casali, B.; Medarametla, L.; Landreth, G.E. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci. Rep. 2017, 7, 42270. [Google Scholar] [CrossRef] [Green Version]
- Mounier, A.; Georgiev, D.; Nam, K.N.; Fitz, N.F.; Castranio, E.L.; Wolfe, C.M.; Cronican, A.A.; Schug, J.; Lefterov, I.; Koldamova, R. Bexarotene-Activated Retinoid X Receptors Regulate Neuronal Differentiation and Dendritic Complexity. J. Neurosci. 2015, 35, 11862–11876. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Nakashima, K.I.; Hirai, T.; Inoue, M. Neuroprotective effect of naturally occurring RXR agonists isolated from Sophora tonkinensis Gagnep. on amyloid-beta-induced cytotoxicity in PC12 cells. J. Nat. Med. 2019, 73, 154–162. [Google Scholar] [CrossRef]
- Wang, W.; Nakashima, K.I.; Hirai, T.; Inoue, M. Anti-inflammatory effects of naturally occurring retinoid X receptor agonists isolated from Sophora tonkinensis Gagnep. via retinoid X receptor/liver X receptor heterodimers. J. Nat. Med. 2019, 73, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Chen, H.K.; Ji, Z.S.; Dodson, S.E.; Miranda, R.D.; Rosenblum, C.I.; Reynolds, I.J.; Freedman, S.B.; Weisgraber, K.H.; Huang, Y.; Mahley, R.W. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem. 2011, 286, 5215–5221. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.K.; Liu, Z.; Meyer-Franke, A.; Brodbeck, J.; Miranda, R.D.; McGuire, J.G.; Pleiss, M.A.; Ji, Z.S.; Balestra, M.E.; Walker, D.W.; et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem. 2012, 287, 5253–5266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodbeck, J.; McGuire, J.; Liu, Z.; Meyer-Franke, A.; Balestra, M.E.; Jeong, D.E.; Pleiss, M.; McComas, C.; Hess, F.; Witter, D.; et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem. 2011, 286, 17217–17226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raulin, A.C.; Kraft, L.; Al-Hilaly, Y.K.; Xue, W.F.; McGeehan, J.E.; Atack, J.R.; Serpell, L. The Molecular Basis for Apolipoprotein E4 as the Major Risk Factor for Late-Onset Alzheimer’s Disease. J. Mol. Biol 2019, 431, 2248–2265. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Li, A.; Xiong, M.; Bien-Ly, N.; Jiang, H.; Zhang, Y.; Finn, M.B.; Hoyle, R.; Keyser, J.; Lefton, K.B.; et al. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J. Clin. Investig. 2018, 128, 2144–2155. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene. Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef]
- Hudry, E.; Dashkoff, J.; Roe, A.D.; Takeda, S.; Koffie, R.M.; Hashimoto, T.; Scheel, M.; Spires-Jones, T.; Arbel-Ornath, M.; Betensky, R.; et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl. Med. 2013, 5, 212ra161. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Gottesdiener, A.J.; Parmar, M.; Li, M.; Kaminsky, S.M.; Chiuchiolo, M.J.; Sondhi, D.; Sullivan, P.M.; Holtzman, D.M.; Crystal, R.G.; et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 2016, 44, 159–172. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Brendza, R.P.; Heuser, J.E.; Kierson, M.; Cirrito, J.R.; Fryer, J.; Sullivan, P.M.; Fagan, A.M.; Han, X.; Holtzman, D.M. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem. Int. 2001, 39, 415–425. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Legleiter, J.; Han, X.; Fryer, J.D.; Kowalewski, T.; Holtzman, D.M. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 2004, 279, 40987–40993. [Google Scholar] [CrossRef] [Green Version]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Kim, J.; Li, A.; Knoten, A.; Jain, S.; Hirsch-Reinshagen, V.; Wellington, C.L.; Bales, K.R.; et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Investig. 2008, 118, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Hirsch-Reinshagen, V.; Zhou, S.; Burgess, B.L.; Bernier, L.; McIsaac, S.A.; Chan, J.Y.; Tansley, G.H.; Cohn, J.S.; Hayden, M.R.; Wellington, C.L. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 2004, 279, 41197–41207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Hartman, R.E.; Bales, K.R.; Paul, S.M.; Holtzman, D.M. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2005, 280, 43236–43242. [Google Scholar] [CrossRef] [Green Version]
- Koldamova, R.; Staufenbiel, M.; Lefterov, I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 2005, 280, 43224–43235. [Google Scholar] [CrossRef] [Green Version]
- Koldamova, R.; Lefterov, I. Role of LXR and ABCA1 in the pathogenesis of Alzheimer’s disease—implications for a new therapeutic approach. Curr. Alzheimer Res. 2007, 4, 171–178. [Google Scholar] [CrossRef]
- Nordestgaard, L.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement. 2015, 11, 1430–1438. [Google Scholar] [CrossRef]
- Yassine, H.N.; Feng, Q.; Chiang, J.; Petrosspour, L.M.; Fonteh, A.N.; Chui, H.C.; Harrington, M.G. ABCA1-Mediated Cholesterol Efflux Capacity to Cerebrospinal Fluid Is Reduced in Patients With Mild Cognitive Impairment and Alzheimer’s Disease. J. Am. Heart Assoc. 2016, 5, e002886. [Google Scholar] [CrossRef] [Green Version]
- Tai, L.M.; Thomas, R.; Marottoli, F.M.; Koster, K.P.; Kanekiyo, T.; Morris, A.W.; Bu, G. The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 2016, 131, 709–723. [Google Scholar] [CrossRef] [Green Version]
- Namjoshi, D.R.; Martin, G.; Donkin, J.; Wilkinson, A.; Stukas, S.; Fan, J.; Carr, M.; Tabarestani, S.; Wuerth, K.; Hancock, R.E.; et al. The liver X receptor agonist GW3965 improves recovery from mild repetitive traumatic brain injury in mice partly through apolipoprotein E. PLoS ONE 2013, 8, e53529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J.L.; Zhong, K.; Kinney, J.W.; Heaney, C.; Moll-Tudla, J.; Joshi, A.; Pontecorvo, M.; Devous, M.; Tang, A.; Bena, J. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene Xin moderate Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [Green Version]
- Jalil, A.; Bourgeois, T.; Menegaut, L.; Lagrost, L.; Thomas, C.; Masson, D. Revisiting the Role of LXRs in PUFA Metabolism and Phospholipid Homeostasis. Int. J. Mol. Sci. 2019, 20, 3787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecht, W.J.; Harris, F.M.; Chang, S.; Tesseur, I.; Yu, G.Q.; Xu, Q.; Dee Fish, J.; Wyss-Coray, T.; Buttini, M.; Mucke, L.; et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci. 2004, 24, 2527–2534. [Google Scholar] [CrossRef]
- Mahley, R.W.; Huang, Y. Small-molecule structure correctors target abnormal protein structure and function: Structure corrector rescue of apolipoprotein E4-associated neuropathology. J. Med. Chem. 2012, 55, 8997–9008. [Google Scholar] [CrossRef] [Green Version]
- Koch, S.; Donarski, N.; Goetze, K.; Kreckel, M.; Stuerenburg, H.J.; Buhmann, C.; Beisiegel, U. Characterization of four lipoprotein classes in human cerebrospinal fluid. J. Lipid Res. 2001, 42, 1143–1151. [Google Scholar]



{kind=link}
{kind=link}
| Isoform | Amino Acids (112, 158) | Structural Description |
|---|---|---|
| ApoE2 | Cys, Cys |
|
| ApoE3 | Cys, Arg |
|
| ApoE4 | Arg, Arg |
|
| Class | Description | Example | References |
|---|---|---|---|
| ABCA1 agonist | Antisense oligonucleotides | miR-33 ARF6 | [104] [105] |
| Small peptides | CS-6253 Ac-hE18A-NH2 4F | [73], [106,107] [108] [73], [109,110,111] | |
| Nuclear Receptor agonist | LXR agonist | TO901317 GW3965 | [112,113,114] [112,113,114] |
| RXR agonist | Bexarotene LG100268 SPF1 and SPF2 | [115,116,117,118,119] [115,116,117] [120,121] | |
| Structure corrector | Small molecule that corrects apoE4 structure | PH002 GIND105 and GIND-25 | [122] [123,124,125] |
| Immunotherapy | Targets non-lipidated apoE4 | HAE-1 and HAE-4 | [126,127] |
| Biologics | AAV-directed therapy | AAV-expressing human APOE2 gene | [84], [128,129,130,131] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanfranco, M.F.; Ng, C.A.; Rebeck, G.W. ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6336. https://doi.org/10.3390/ijms21176336
Lanfranco MF, Ng CA, Rebeck GW. ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(17):6336. https://doi.org/10.3390/ijms21176336
Chicago/Turabian StyleLanfranco, Maria Fe, Christi Anne Ng, and G. William Rebeck. 2020. "ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 17: 6336. https://doi.org/10.3390/ijms21176336
APA StyleLanfranco, M. F., Ng, C. A., & Rebeck, G. W. (2020). ApoE Lipidation as a Therapeutic Target in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(17), 6336. https://doi.org/10.3390/ijms21176336