CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene, and Pathophysiological Conditions

 ,
,  , ,
, ,  , , ,
, , ,  and
and
Abstract
:
1. Introduction
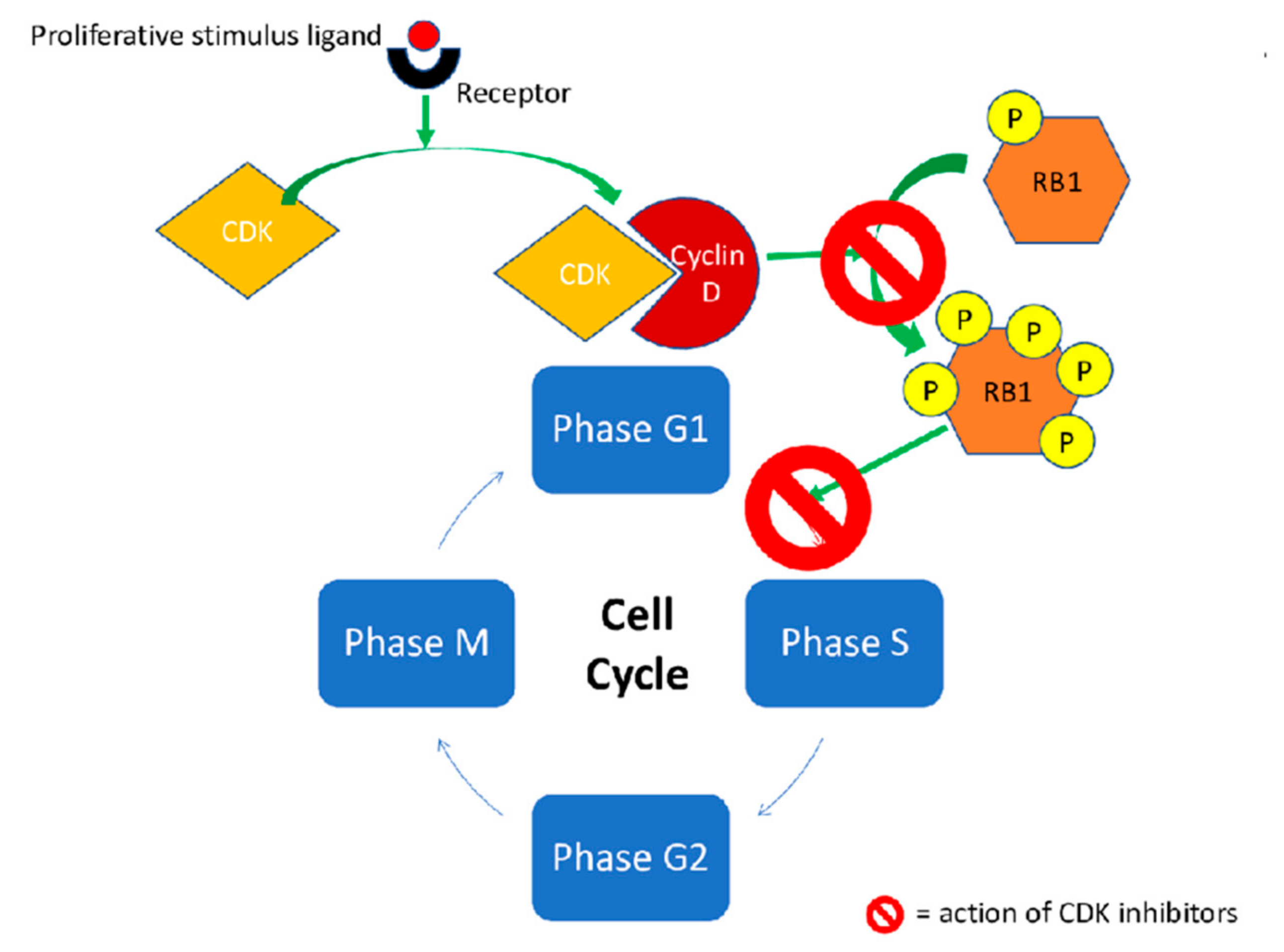
1.1. Pharmacodynamics
1.2. Safety
1.3. Pharmacokinetics
2. Potential Drug-Drug Interactions
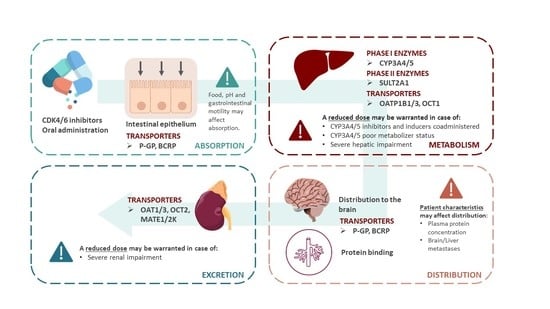
2.1. Potential Drug-Drug Interactions with ADME
2.1.1. Agents That May Alter CDK4/6 Inhibitors Absorption
- (1)
- Gastric pH Elevating Medications
- (2)
- Membrane Transporters
2.1.2. Agents That May Alter CDK4/6 Inhibitors Distribution
2.1.3. Agents That May Alter CDK4/6 Inhibitors Metabolism
- (1)
- CYP3A Inhibitors May Increase CDK4/6 Inhibitors Plasma Concentrations
- (2)
- CYP3A Inducers May Decrease CDK4/6 Inhibitors Plasma Concentrations
2.1.4. Agents That May Be Altered by Co-Administration with CDK4/6 Inhibitors
2.1.5. Pain Killer (Opioids and NSAIDs)
2.2. Potential Drug-Drug Interactions with Non ADME Agents That May Potentiate CDK4/6 Inhibitors Toxicity
Antidepressant
2.3. Other DDIs
2.3.1. Osteoporosis Treatment (Denosumab, Vit D)
2.3.2. Potential SULT2A1-Mediated DDIs with CDK4/6 Inhibitors
3. Potential Drug-Gene Interactions with ADME
3.1. Phase I Enzymes: CYP3A4 and CYP3A5
3.2. Phase II Enzymes: SULTs
3.3. Impact of Genes with Indirect Impact on CYP3A Activity
3.4. Transporters (ABCB1 and ABCG2)
4. Phenoconversion
5. Potential Drug-Pathophysiological Interactions
5.1. Hepatic Impairment
5.2. Renal Impairment
5.3. Gender and Hormonal Status
5.4. Inflammation and Cancer
5.5. Brain Metastases from Breast Cancer
5.6. Obesity
5.7. Age
6. Mechanisms of Resistance to CDKIs
7. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonelli, M.; La Monica, S.; Fumarola, C.; Alfieri, R. Multiple effects of CDK4/6 inhibition in cancer: From cell cycle arrest to immunomodulation. Biochem. Pharmacol. 2019, 170, 113676. [Google Scholar] [CrossRef] [PubMed]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: Past, present, and future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef]
- Kotake, T.; Toi, M. Abemaciclib for the treatment of breast cancer. Expert. Opin. Pharm. 2018, 19, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A. Molecular pathways: CDK4 inhibitors for cancer therapy. Clin. Cancer Res. 2014, 20, 3379–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.J.; Cheng, J.; Bloomquist, E.; Sanchez, J.; Wedam, S.B.; Singh, H.; Amiri-Kordestani, L.; Ibrahim, A.; Sridhara, R.; Goldberg, K.B.; et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: A US Food and Drug Administration pooled analysis. Lancet Oncol. 2020, 21, 250–260. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.-C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- KISQALI® (ribociclib). Highlights of Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/209092s004lbl.pdf (accessed on 14 August 2020).
- Committee for Medicinal Products for Human Use (CHMP). KISQALI® (ribociclib) EPAR Assessment Report Variation. Available online: https://www.ema.europa.eu/en/documents/variation-report/kisqali-h-c-4213-ii-0004-epar-assessment-report-variation_en.pdf (accessed on 31 August 2020).
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.-A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. 2019, 6, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR+/HER2- Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef] [Green Version]
- Committee for Medicinal Products for Human Use (CHMP). VERZENIOTM (abemaciclib) EPAR Assessment Report. Available online: https://www.ema.europa.eu/en/documents/assessment-report/verzenios-epar-public-assessment-report_en.pdf (accessed on 31 August 2020).
- Petrelli, F.; Ghidini, A.; Pedersini, R.; Cabiddu, M.; Borgonovo, K.; Parati, M.C.; Ghilardi, M.; Amoroso, V.; Berruti, A.; Barni, S. Comparative efficacy of palbociclib, ribociclib and abemaciclib for ER+ metastatic breast cancer: An adjusted indirect analysis of randomized controlled trials. Breast Cancer Res. Treat. 2019, 174, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.C.; Lord, S.; Finn, R.S.; Lim, E.; Martin, A.; Loi, S.; Lynch, J.; Friedlander, M.; Lee, C.K. The impact of ethnicity on efficacy and toxicity of cyclin D kinase 4/6 inhibitors in advanced breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2019, 174, 271–278. [Google Scholar] [CrossRef]
- Pala, L.; Conforti, F.; Goldhirsch, A. Ethnicity-based differences in breast cancer features and responsiveness to CDK4/6 inhibitors combined with endocrine therapy. Lancet Oncol. 2020, 21, 130. [Google Scholar] [CrossRef]
- Fukunaga, S.; Kusama, M.; Arnold, F.L.; Ono, S. Ethnic differences in pharmacokinetics in new drug applications and approved doses in Japan. J. Clin. Pharm. 2011, 51, 1237–1240. [Google Scholar] [CrossRef]
- Arnold, F.L.; Kusama, M.; Ono, S. Exploring differences in drug doses between Japan and Western countries. Clin. Pharmacol. Ther. 2010, 87, 714–720. [Google Scholar] [CrossRef]
- Warth, B.; Raffeiner, P.; Granados, A.; Huan, T.; Fang, M.; Forsberg, E.M.; Benton, H.P.; Goetz, L.; Johnson, C.H.; Siuzdak, G. Metabolomics Reveals that Dietary Xenoestrogens Alter Cellular Metabolism Induced by Palbociclib/Letrozole Combination Cancer Therapy. Cell Chem. Biol. 2018, 25, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Kan, Z.; Ding, Y.; Kim, J.; Jung, H.H.; Chung, W.; Lal, S.; Cho, S.; Fernandez-Banet, J.; Lee, S.K.; Kim, S.W.; et al. Multi-omics profiling of younger Asian breast cancers reveals distinctive molecular signatures. Nat. Commun. 2018, 9, 1725. [Google Scholar] [CrossRef]
- Doi, T.; Hewes, B.; Kakizume, T.; Tajima, T.; Ishikawa, N.; Yamada, Y. Phase I study of single-agent ribociclib in Japanese patients with advanced solid tumors. Cancer Sci. 2018, 109, 193–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.R.; Smith, R.L. Addressing phenoconversion: The Achilles’ heel of personalized medicine. Br. J. Clin. Pharm. 2015, 79, 222–240. [Google Scholar] [CrossRef] [Green Version]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, M.E.; Corsino, P.E.; Narayan, S.; Law, B.K. Cyclin-Dependent Kinase Inhibitors as Anticancer Therapeutics. Mol. Pharmacol. 2015, 88, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Bloomquist, E.; Tang, S.; Fu, W.; Bi, Y.; Liu, Q.; Yu, J.; Zhao, P.; Palmby, T.R.; Goldberg, K.B.; et al. FDA Approval: Ribociclib for the Treatment of Postmenopausal Women with Hormone Receptor-Positive, HER2-Negative Advanced or Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Sammons, S.L.; Topping, D.L.; Blackwell, K.L. HR+, HER2- Advanced Breast Cancer and CDK4/6 Inhibitors: Mode of Action, Clinical Activity, and Safety Profiles. Curr. Cancer Drug Targets 2017, 17, 637–649. [Google Scholar] [CrossRef]
- Poratti, M.; Marzaro, G. Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153. [Google Scholar] [CrossRef]
- Hafner, M.; Mills, C.E.; Subramanian, K.; Chen, C.; Chung, M.; Boswell, S.A.; Everley, R.A.; Liu, C.; Walmsley, C.S.; Juric, D.; et al. Multi-omics profiling establishes the polypharmacology of FDA Approved CDK4/6 inhibitors and the potential for differential clinical activity. Cell Chem. Biol. 2019, 26, 1067–1080. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.-K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Wood, D.J.; Endicott, J.A. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.; Thijssen, B.; McDermott, U.; Garnett, M.; Wessels, L.F.A.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Committee for Medicinal Products for Human Use (CHMP). IBRANCE® (palbociclib) EPAR Assessment Report. Available online: https://www.ema.europa.eu/en/documents/assessment-report/piqray-epar-public-assessment-report_en.pdf (accessed on 31 August 2020).
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, M.G.; Deshpande, A.; Schlichting, N.; Hinds, E.A.; Mao, C.; Dose, M.; Hu, G.; Van Etten, R.A.; Gounari, F.; Hinds, P.W. CDK6 kinase activity is required for thymocyte development. Blood 2011, 117, 6120–6131. [Google Scholar] [CrossRef]
- Garriga, J.; Graña, X. CDK9 inhibition strategy defines distinct sets of target genes. BMC Res. Notes 2014, 7, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathy, D.; Hortobagyi, G.N.; Chan, A.; Im, S.-A.; Chia, S.; Yardley, D.; Esteva, F.J.; Hurvitz, S.A.; Ridolfi, A.; Slamon, D. Pooled safety analysis of first-line ribociclib (rib) plus endocrine therapy (ET) in HR+/HER2– advanced breast cancer (ABC). Ann. Oncol. 2019, 30, 47–64. [Google Scholar] [CrossRef]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.-A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: A randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5. [Google Scholar] [CrossRef] [Green Version]
- Diéras, V.; Rugo, H.S.; Schnell, P.; Gelmon, K.; Cristofanilli, M.; Loi, S.; Colleoni, M.; Lu, D.R.; Mori, A.; Gauthier, E.; et al. Long-term Pooled Safety Analysis of Palbociclib in Combination with Endocrine Therapy for HR+/HER2- Advanced Breast Cancer. J. Natl. Cancer Inst. 2018, 111, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Thein, K.Z.; Htut, T.W.; Ball, S.; Swarup, S.; Sultan, A.; Oo, T.H. Venous thromboembolism risk in patients with hormone receptor-positive HER2-negative metastatic breast cancer treated with combined CDK 4/6 inhibitors plus endocrine therapy versus endocrine therapy alone: A systematic review and meta-analysis of randomized controlled trials. Breast Cancer Res. Treat. 2020, 183, 479–487. [Google Scholar] [CrossRef]
- Undevia, S.D.; Gomez-Abuin, G.; Ratain, M.J. Pharmacokinetic variability of anticancer agents. Nat. Rev. Cancer 2005, 5, 447–458. [Google Scholar] [CrossRef]
- Schmidt, M. Palbociclib—From Bench to Bedside and Beyond. Breast Care (Basel) 2016, 11, 177–181. [Google Scholar] [CrossRef] [Green Version]
- IBRANCE® (palbociclib). Highlights of Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/207103s008lbl.pdf (accessed on 11 August 2020).
- FDA. VERZENIOTM (abemaciclib). Highlights of Prescribing Information. 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208855s000lbl.pdf (accessed on 14 August 2020).
- Chen, P.; Lee, N.V.; Hu, W.; Xu, M.; Ferre, R.A.; Lam, H.; Bergqvist, S.; Solowiej, J.; Diehl, W.; He, Y.-A.; et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol. Cancer Ther. 2016, 15, 2273–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; LoRusso, P.M.; DeMichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, Dose-Escalation Trial of the Oral Cyclin-Dependent Kinase 4/6 Inhibitor PD 0332991, Administered Using a 21-Day Schedule in Patients with Advanced Cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S.; et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res. 2016, 22, 5696–5705. [Google Scholar] [CrossRef] [Green Version]
- Bellet, M.; Ahmad, F.; Villanueva, R.; Valdivia, C.; Palomino-Doza, J.; Ruiz, A.; Gonzàlez, X.; Adrover, E.; Azaro, A.; Valls-Margarit, M.; et al. Palbociclib and ribociclib in breast cancer: Consensus workshop on the management of concomitant medication. Adv. Med. Oncol 2019, 11, 1758835919833867. [Google Scholar] [CrossRef]
- Raub, T.J.; Wishart, G.N.; Kulanthaivel, P.; Staton, B.A.; Ajamie, R.T.; Sawada, G.A.; Gelbert, L.M.; Shannon, H.E.; Sanchez-Martinez, C.; De Dios, A. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab. Dispos. 2015, 43, 1360–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, Y.T.; Davis, A.; Baker, S.J.; Campagne, O.; Stewart, C.F. CNS penetration of the CDK4/6 inhibitor ribociclib in non-tumor bearing mice and mice bearing pediatric brain tumors. Cancer Chemother. Pharmacol. 2019, 84, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Chávez, A.; van Hoppe, S.; Rosing, H.; Lebre, M.C.; Tibben, M.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein Limits Ribociclib Brain Exposure and CYP3A4 Restricts Its Oral Bioavailability. Mol. Pharm. 2019, 16, 3842–3852. [Google Scholar] [CrossRef]
- Chong, Q.-Y.; Kok, Z.-H.; Bui, N.-L.-C.; Xiang, X.; Wong, A.L.-A.; Yong, W.-P.; Sethi, G.; Lobie, P.E.; Wang, L.; Goh, B.-C. A unique CDK4/6 inhibitor: Current and future therapeutic strategies of abemaciclib. Pharmacol. Res. 2020, 156, 104686. [Google Scholar] [CrossRef]
- Rascon, K.; Flajc, G.; De Angelis, C.; Liu, X.; Trivedi, M.V.; Ekinci, E. Ribociclib in HR+/HER2- Advanced or Metastatic Breast Cancer Patients. Ann. Pharm. 2019, 53, 501–509. [Google Scholar] [CrossRef]
- Ruiz-Garcia, A.; Plotka, A.; O’Gorman, M.; Wang, D.D. Effect of food on the bioavailability of palbociclib. Cancer Chemother. Pharmacol. 2017, 79, 527–533. [Google Scholar] [CrossRef]
- Tate, S.C.; Sykes, A.K.; Kulanthaivel, P.; Chan, E.M.; Turner, P.K.; Cronier, D.M. A Population Pharmacokinetic and Pharmacodynamic Analysis of Abemaciclib in a Phase I Clinical Trial in Cancer Patients. Clin. Pharm. 2018, 57, 335–344. [Google Scholar] [CrossRef]
- Curigliano, G.; Criscitiello, C.; Esposito, A.; Intra, M.; Minucci, S. Pharmacokinetic drug evaluation of ribociclib for the treatment of metastatic, hormone-positive breast cancer. Expert Opin. Drug Metab. Toxicol. 2017, 13, 575–581. [Google Scholar] [CrossRef]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Sonke, G.S.; Hart, L.L.; Campone, M.; Erdkamp, F.; Janni, W.; Verma, S.; Villanueva, C.; Jakobsen, E.; Alba, E.; Wist, E.; et al. Ribociclib with letrozole vs. letrozole alone in elderly patients with hormone receptor-positive, HER2-negative breast cancer in the randomized MONALEESA-2 trial. Breast Cancer Res. Treat. 2018, 167, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Wang, X. Role of vitamin D receptor in the regulation of CYP3A gene expression. Acta Pharm. Sin. B 2019, 9, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- James, M.O.; Ambadapadi, S. Interactions of cytosolic sulfotransferases with xenobiotics. Drug Metab. Rev. 2013, 45, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Marto, N.; Morello, J.; Monteiro, E.C.; Pereira, S.A. Implications of sulfotransferase activity in interindividual variability in drug response: Clinical perspective on current knowledge. Drug Metab. Rev. 2017, 49, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Fukushima-Uesaka, H.; Saito, Y.; Watanabe, H.; Shiseki, K.; Saeki, M.; Nakamura, T.; Kurose, K.; Sai, K.; Komamura, K.; Ueno, K.; et al. Haplotypes of CYP3A4 and their close linkage with CYP3A5 haplotypes in a Japanese population. Hum. Mutat. 2004, 23, 100. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef]
- Park, J.-E.; Kim, K.-B.; Bae, S.K.; Moon, B.-S.; Liu, K.-H.; Shin, J.-G. Contribution of cytochrome P450 3A4 and 3A5 to the metabolism of atorvastatin. Xenobiotica 2008, 38, 1240–1251. [Google Scholar] [CrossRef]
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharm. J. 2011, 11, 274–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, S.D.; Teft, W.A.; Choi, Y.-H.; Winquist, E.; Kim, R.B. Tamoxifen-associated hot flash severity is inversely correlated with endoxifen concentration and CYP3A4*22. Breast Cancer Res. Treat. 2014, 145, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Gong, I.Y.; Dingle, B.; Potvin, K.; Younus, J.; Vandenberg, T.A.; Brackstone, M.; Perera, F.E.; Choi, Y.-H.; Zou, G.; et al. CYP3A4 and seasonal variation in vitamin D status in addition to CYP2D6 contribute to therapeutic endoxifen level during tamoxifen therapy. Breast Cancer Res. Treat. 2013, 139, 95–105. [Google Scholar] [CrossRef]
- Diekstra, M.H.M.; Klümpen, H.J.; Lolkema, M.P.J.K.; Yu, H.; Kloth, J.S.L.; Gelderblom, H.; van Schaik, R.H.N.; Gurney, H.; Swen, J.J.; Huitema, A.D.R.; et al. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin. Pharmacol. Ther. 2014, 96, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Kreutz, R.P.; Owens, J.; Jin, Y.; Nystrom, P.; Desta, Z.; Kreutz, Y.; Breall, J.A.; Li, L.; Chiang, C.; Kovacs, R.J.; et al. Cytochrome P450 3A4*22, PPAR-α, and ARNT polymorphisms and clopidogrel response. Clin. Pharm. 2013, 5, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Li, Y.; Tang, J.; Zhang, J.; Zou, Y.; Cai, B.; Wang, L. Influence of CYP3A4, CYP3A5 and MDR-1 polymorphisms on tacrolimus pharmacokinetics and early renal dysfunction in liver transplant recipients. Gene 2013, 512, 226–231. [Google Scholar] [CrossRef]
- Miao, J.; Jin, Y.; Marunde, R.L.; Gorski, C.J.; Kim, S.; Quinney, S.; Radovich, M.; Li, L.; Hall, S.D. Association of genotypes of the CYP3A cluster with midazolam disposition In Vivo. Pharm. J. 2009, 9, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Tremmel, R.; Klein, K.; Battke, F.; Fehr, S.; Winter, S.; Scheurenbrand, T.; Schaeffeler, E.; Biskup, S.; Schwab, M.; Zanger, U.M. Copy number variation profiling in pharmacogenes using panel-based exome resequencing and correlation to human liver expression. Hum. Genet. 2020, 139, 137–149. [Google Scholar] [CrossRef]
- Gordon, A.S.; Tabor, H.K.; Johnson, A.D.; Snively, B.M.; Assimes, T.L.; Auer, P.L.; Ioannidis, J.P.A.; Peters, U.; Robinson, J.G.; Sucheston, L.E.; et al. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug-metabolism genes in a large-scale exome dataset. Hum. Mol. Genet. 2014, 23, 1957–1963. [Google Scholar] [CrossRef] [Green Version]
- Ingelman-Sundberg, M.; Mkrtchian, S.; Zhou, Y.; Lauschke, V.M. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum. Genom. 2018, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Westlind-Johnsson, A.; Hermann, R.; Huennemeyer, A.; Hauns, B.; Lahu, G.; Nassr, N.; Zech, K.; Ingelman-Sundberg, M.; von Richter, O. Identification and characterization of CYP3A4*20, a novel rare CYP3A4 allele without functional activity. Clin. Pharmacol. Ther. 2006, 79, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Werk, A.N.; Lefeldt, S.; Bruckmueller, H.; Hemmrich-Stanisak, G.; Franke, A.; Roos, M.; Küchle, C.; Steubl, D.; Schmaderer, C.; Bräsen, J.H.; et al. Identification and characterization of a defective CYP3A4 genotype in a kidney transplant patient with severely diminished tacrolimus clearance. Clin. Pharmacol. Ther. 2014, 95, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Birdwell, K.A.; Decker, B.; Barbarino, J.M.; Peterson, J.F.; Stein, C.M.; Sadee, W.; Wang, D.; Vinks, A.A.; He, Y.; Swen, J.J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef] [Green Version]
- García-Anguita, A.; Ortega, L.; Garcés, C. Relationship between polymorphisms in the sulfotransferase SULT2A1 gene and dehydroepiandrosterone sulfate concentration in children. Exp. Biol. Med. (Maywood) 2013, 238, 163–166. [Google Scholar] [CrossRef]
- Schulze, J.; Johansson, M.; Thörngren, J.-O.; Garle, M.; Rane, A.; Ekström, L. SULT2A1 Gene Copy Number Variation is Associated with Urinary Excretion Rate of Steroid Sulfates. Front. Endocrinol. (Lausanne) 2013, 4, 88. [Google Scholar] [CrossRef] [Green Version]
- Drocourt, L.; Ourlin, J.-C.; Pascussi, J.-M.; Maurel, P.; Vilarem, M.-J. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem. 2002, 277, 25125–25132. [Google Scholar] [CrossRef] [Green Version]
- De Mattia, E.; Cecchin, E.; Roncato, R.; Toffoli, G. Pregnane X receptor, constitutive androstane receptor and hepatocyte nuclear factors as emerging players in cancer precision medicine. Pharmacogenomics 2016, 17, 1547–1571. [Google Scholar] [CrossRef]
- Elens, L.; Capron, A.; van Schaik, R.H.N.; De Meyer, M.; De Pauw, L.; Eddour, D.C.; Latinne, D.; Wallemacq, P.; Mourad, M.; Haufroid, V. Impact of CYP3A4*22 allele on tacrolimus pharmacokinetics in early period after renal transplantation: Toward updated genotype-based dosage guidelines. Drug Monit. 2013, 35, 608–616. [Google Scholar] [CrossRef]
- Leschziner, G.; Zabaneh, D.; Pirmohamed, M.; Owen, A.; Rogers, J.; Coffey, A.J.; Balding, D.J.; Bentley, D.B.; Johnson, M.R. Exon sequencing and high resolution haplotype analysis of ABC transporter genes implicated in drug resistance. Pharmacogenet. Genom. 2006, 16, 439–450. [Google Scholar] [CrossRef]
- Salama, N.N.; Yang, Z.; Bui, T.; Ho, R.J.Y. MDR1 haplotypes significantly minimize intracellular uptake and transcellular P-gp substrate transport in recombinant LLC-PK1 cells. J. Pharm. Sci. 2006, 95, 2293–2308. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Wakita, H.; Miura, M.; Scott, S.A.; Nishii, K.; Masuko, M.; Sakai, M.; Maeda, Y.; Ishige, K.; Kashimura, M.; et al. Correlation between imatinib pharmacokinetics and clinical response in Japanese patients with chronic-phase chronic myeloid leukemia. Clin. Pharmacol. Ther. 2010, 88, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cusatis, G.; Brahmer, J.; Sparreboom, A.; Robey, R.W.; Bates, S.E.; Hidalgo, M.; Baker, S.D. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol. Ther. 2007, 6, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Terada, T.; Kamba, T.; Fukudo, M.; Katsura, T.; Nakamura, E.; Ogawa, O.; Inui, K. ABCG2 421C>A polymorphism and high exposure of sunitinib in a patient with renal cell carcinoma. Ann. Oncol. 2010, 21, 1382–1383. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Hirose, T.; Kusumoto, S.; Sugiyama, T.; Shirai, T.; Nakashima, M.; Akiyama, Y.; Sasaki, Y. High exposure to erlotinib and severe drug-induced interstitial lung disease in patients with non-small-cell lung cancer. Lung Cancer 2014, 86, 113–114. [Google Scholar] [CrossRef]
- Rudin, C.M.; Liu, W.; Desai, A.; Karrison, T.; Jiang, X.; Janisch, L.; Das, S.; Ramirez, J.; Poonkuzhali, B.; Schuetz, E.; et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J. Clin. Oncol. 2008, 26, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Reyner, E.; Lum, B.; Jing, J.; Kagedal, M.; Ware, J.A.; Dickmann, L.J. Intrinsic and Extrinsic Pharmacokinetic Variability of Small Molecule Targeted Cancer Therapy. Clin. Transl. Sci. 2020, 13, 410–418. [Google Scholar] [CrossRef]
- Kelly, C.M.; Juurlink, D.N.; Gomes, T.; Duong-Hua, M.; Pritchard, K.I.; Austin, P.C.; Paszat, L.F. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: A population based cohort study. BMJ 2010, 340, c693. [Google Scholar] [CrossRef] [Green Version]
- Schaffner, F.; Poper, H. Capillarization of hepatic sinusoids in man. Gastroenterology 1963, 44, 239–242. [Google Scholar] [CrossRef]
- Van Beers, B.E.; Materne, R.; Annet, L.; Hermoye, L.; Sempoux, C.; Peeters, F.; Smith, A.M.; Jamart, J.; Horsmans, Y. Capillarization of the sinusoids in liver fibrosis: Noninvasive assessment with contrast-enhanced MRI in the rabbit. Magn. Reason. Med. 2003, 49, 692–699. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Saxena, R.; Storniolo, A.M.V.; Chalasani, N. Pseudocirrhosis and liver failure in patients with metastatic breast cancer after treatment with palbociclib. Hepatology 2017, 65, 1762–1764. [Google Scholar] [CrossRef] [PubMed]
- Schlotman, A.; Stater, A.; Schuler, K.; Heideman, J.; Abramson, V. Grade 3 Hepatotoxicity following Fulvestrant, Palbociclib, and Erdafitinib Therapy in a Patient with ER-Positive/PR-Negative/HER2-Negative Metastatic Breast Cancer: A Case Report. Case Rep. Oncol. 2020, 13, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Atallah, R.; Parker, N.A.; Hamouche, K.; Truong, Q.V.; Dingwall, M. Palbociclib-Induced Liver Failure. Kans J. Med. 2020, 13, 81–82. [Google Scholar] [CrossRef] [PubMed]
- EMA. Kisqali Procedural Steps Taken and Scientific Information after the Authorisation 2020. Available online: https://www.ema.europa.eu/en/documents/procedural-steps-after/kisqali-epar-procedural-steps-taken-scientific-information-after-authorisation_en.pdf (accessed on 31 August 2020).
- Kim, E.S. Abemaciclib: First Global Approval. Drugs 2017, 77, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Freire, A.C.; Basit, A.W.; Choudhary, R.; Piong, C.W.; Merchant, H.A. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. Int. J. Pharm. 2011, 415, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Samant, T.S.; Dhuria, S.; Lu, Y.; Laisney, M.; Yang, S.; Grandeury, A.; Mueller-Zsigmondy, M.; Umehara, K.; Huth, F.; Miller, M.; et al. Ribociclib Bioavailability Is Not Affected by Gastric pH Changes or Food Intake: In Silico and Clinical Evaluations. Clin. Pharmacol. Ther. 2018, 104, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, M.F.; Ludington, S.S.; Chen, M.-L.; Stewart, P.W.; Huang, S.-M.; Watkins, P.B. Do men and women differ in proximal small intestinal CYP3A or P-glycoprotein expression? Drug Metab. Dispos. 2005, 33, 426–433. [Google Scholar] [CrossRef] [Green Version]
- MacLean, C.; Moenning, U.; Reichel, A.; Fricker, G. Closing the gaps: A full scan of the intestinal expression of p-glycoprotein, breast cancer resistance protein, and multidrug resistance-associated protein 2 in male and female rats. Drug Metab. Dispos. 2008, 36, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Wolbold, R.; Klein, K.; Burk, O.; Nüssler, A.K.; Neuhaus, P.; Eichelbaum, M.; Schwab, M.; Zanger, U.M. Sex is a major determinant of CYP3A4 expression in human liver. Hepatology 2003, 38, 978–988. [Google Scholar] [CrossRef]
- Merino, G.; van Herwaarden, A.E.; Wagenaar, E.; Jonker, J.W.; Schinkel, A.H. Sex-dependent expression and activity of the ATP-binding cassette transporter breast cancer resistance protein (BCRP/ABCG2) in liver. Mol. Pharmacol. 2005, 67, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.R.; Fromm, B.S.; Steinman, R.T.; Meissner, M.D.; Lehmann, M.H. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA 1993, 270, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, I.; Kilborn, M.J.; Liu, X.K.; Pezzullo, J.C.; Woosley, R.L. Drug-induced QT prolongation in women during the menstrual cycle. JAMA 2001, 285, 1322–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, S.C.; Smith, R.L.; Waring, R.H. The menstrual cycle and drug metabolism. Curr. Drug Metab. 2009, 10, 499–507. [Google Scholar] [CrossRef]
- Ximenez, J.P.B.; de Andrade, J.M.; Marques, M.P.; Coelho, E.B.; Suarez-Kurtz, G.; Lanchote, V.L. Hormonal status affects plasma exposure of tamoxifen and its main metabolites in tamoxifen-treated breast cancer patients. BMC Pharm. Toxicol. 2019, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- Kanakis, G.A.; Jørgensen, N.; Goulis, D.G. Breast Cancer in Men. N. Engl. J. Med. 2018, 379, 1385. [Google Scholar] [CrossRef]
- Frisone, D.; Charrier, M.; Clement, S.; Christinat, Y.; Thouvenin, L.; Homicsko, K.; Michielin, O.; Bodmer, A.; Chappuis, P.O.; McKee, T.A.; et al. Durable response to palbociclib and letrozole in ovarian cancer with CDKN2A loss. Cancer Biol. 2019, 21, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Morgan, E.T.; Goralski, K.B.; Piquette-Miller, M.; Renton, K.W.; Robertson, G.R.; Chaluvadi, M.R.; Charles, K.A.; Clarke, S.J.; Kacevska, M.; Liddle, C.; et al. Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab. Dispos. 2008, 36, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Slaviero, K.A.; Clarke, S.J.; Rivory, L.P. Inflammatory response: An unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol. 2003, 4, 224–232. [Google Scholar] [CrossRef]
- Garattini, S. Pharmacokinetics in cancer chemotherapy. Eur. J. Cancer 2007, 43, 271–282. [Google Scholar] [CrossRef]
- Abdel-Razzak, Z.; Loyer, P.; Fautrel, A.; Gautier, J.C.; Corcos, L.; Turlin, B.; Beaune, P.; Guillouzo, A. Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol. Pharmacol. 1993, 44, 707–715. [Google Scholar]
- Aitken, A.E.; Morgan, E.T. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 2007, 35, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Sunman, J.A.; Hawke, R.L.; LeCluyse, E.L.; Kashuba, A.D.M. Kupffer cell-mediated IL-2 suppression of CYP3A activity in human hepatocytes. Drug Metab. Dispos. 2004, 32, 359–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, N.; Kataoka, H.; Fujino, H.; Nishikawa, J.; Kugawa, F. Different expression patterns of hepatic cytochrome P450 s during anaphylactic or lipopolysaccharide-induced inflammation. Pharmazie 2014, 69, 142–147. [Google Scholar] [PubMed]
- Morgan, E.T.; Dempsey, J.L.; Mimche, S.M.; Lamb, T.J.; Kulkarni, S.; Cui, J.Y.; Jeong, H.; Slitt, A.L. Physiological Regulation of Drug Metabolism and Transport: Pregnancy, Microbiome, Inflammation, Infection, and Fasting. Drug Metab. Dispos. 2018, 46, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivory, L.P.; Slaviero, K.A.; Clarke, S.J. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br. J. Cancer 2002, 87, 277–280. [Google Scholar] [CrossRef]
- Noll, E.M.; Eisen, C.; Stenzinger, A.; Espinet, E.; Muckenhuber, A.; Klein, C.; Vogel, V.; Klaus, B.; Nadler, W.; Rösli, C.; et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat. Med. 2016, 22, 278–287. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Baumann, F.; Knüpfer, H.; Brauckhoff, M.; Horn, L.-C.; Schönfelder, M.; Köhler, U.; Preiss, R. CYP3A4, CYP2C9 and CYP2B6 expression and ifosfamide turnover in breast cancer tissue microsomes. Br. J. Cancer 2004, 90, 911–916. [Google Scholar] [CrossRef] [Green Version]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Kim, M.S.; Shigenaga, J.; Moser, A.; Grunfeld, C.; Feingold, K.R. Suppression of DHEA sulfotransferase (Sult2A1) during the acute-phase response. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E731–E738. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.U.; Amiri-Kordestani, L.; Palmieri, D.; Liewehr, D.J.; Steeg, P.S. CNS metastases in breast cancer: Old challenge, new frontiers. Clin. Cancer Res. 2013, 19, 6404–6418. [Google Scholar] [CrossRef] [Green Version]
- Larochelle, C.; Alvarez, J.I.; Prat, A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011, 585, 3770–3780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, J.W.; Cordon-Cardo, C.; Posner, J.B. P-glycoprotein expression in brain tumors. J. Neuro-Oncol. 1992, 14, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.K.; Le Rhun, E.; Bachelot, T.D.; Yardley, D.A.; Awada, A.; Conte, P.F.; Kabos, P.; Bear, M.; Yang, Z.; Chen, Y.; et al. A phase II study of abemaciclib in patients (pts) with brain metastases (BM) secondary to HR+, HER2− metastatic breast cancer (MBC). J. Clin. Oncol. 2019, 37, 1017. [Google Scholar] [CrossRef]
- Tien, A.-C.; Li, J.; Bao, X.; Derogatis, A.; Kim, S.; Mehta, S.; Sanai, N. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin. Cancer Res. 2019, 25, 5777–5786. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.; Cohen, J.; Wang, N.; Lee, E.; Ligibel, J.; Chukwueke, U.; Keeley, M.; Oh, K.; Whie, M.; Gerstner, E.; et al. A phase II study of palbociclib in progressive brain metastases harboring alterations in the CDK pathway. In Proceedings of the 4th Annual Meeting and Education Day, Phoenix, AZ, USA, 20–24 November 2019; Volume 21, (Neuro-Oncology). p. vi58. [Google Scholar] [CrossRef]
- Bloomgarden, Z.T. Second World Congress on the Insulin Resistance Syndrome: Insulin resistance syndrome and nonalcoholic fatty liver disease. Diabetes Care 2005, 28, 1518–1523. [Google Scholar] [CrossRef] [Green Version]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res. Treat. 2010, 123, 627–635. [Google Scholar] [CrossRef]
- Amadou, A.; Ferrari, P.; Muwonge, R.; Moskal, A.; Biessy, C.; Romieu, I.; Hainaut, P. Overweight, obesity and risk of premenopausal breast cancer according to ethnicity: A systematic review and dose-response meta-analysis. Obes. Rev. 2013, 14, 665–678. [Google Scholar] [CrossRef]
- Begum, P.; Richardson, C.E.; Carmichael, A.R. Obesity in post menopausal women with a family history of breast cancer: Prevalence and risk awareness. Int. Semin. Surg. Oncol. 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, N.M.; Yaghan, R.J.; Abdo, N.M.; Matalka, I.I.; Akhu-Zaheya, L.M.; Al-Mohtaseb, A.H. Impact of Obesity on Clinicopathologic Characteristics and Disease Prognosis in Pre- and Postmenopausal Breast Cancer Patients: A Retrospective Institutional Study. J. Obes. 2019, 2019, 3820759. [Google Scholar] [CrossRef] [Green Version]
- Committee for Human Medicinal Products (CHMP). Reflection Paper on Investigation of Pharmacokinetics and Pharmacodynamics in the Obese Population. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-investigation-pharmacokinetics-pharmacodynamics-obese-population_en.pdf (accessed on 31 August 2020).
- Evans, M.A.; Triggs, E.J.; Cheung, M.; Broe, G.A.; Creasey, H. Gastric emptying rate in the elderly: Implications for drug therapy. J. Am. Geriatr Soc. 1981, 29, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Rubinow, D.R.; Moore, M. Sex-dependent modulation of treatment response. Dialogues Clin. Neurosci. 2004, 6, 39–51. [Google Scholar] [PubMed]
- Robertson, D.R.; Waller, D.G.; Renwick, A.G.; George, C.F. Age-related changes in the pharmacokinetics and pharmacodynamics of nifedipine. Br. J. Clin. Pharm. 1988, 25, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, D.J.; Harmatz, J.S.; Shapiro, L.; Engelhardt, N.; Gouthro, T.A.; Shader, R.I. Sensitivity to triazolam in the elderly. N. Engl. J. Med. 1991, 324, 1691–1698. [Google Scholar] [CrossRef]
- Evans, C. Malnutrition in the elderly: A multifactorial failure to thrive. Perm. J. 2005, 9, 38–41. [Google Scholar] [CrossRef]
- van Assema, D.M.E.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.I.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135, 181–189. [Google Scholar] [CrossRef]
- Tesarova, P. Breast cancer in the elderly-Should it be treated differently? Rep. Pr. Oncol. Radiother. 2012, 18, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Portman, N.; Alexandrou, S.; Carson, E.; Wang, S.; Lim, E.; Caldon, C.E. Overcoming CDK4/6 inhibitor resistance in ER-positive breast cancer. Endocr. Relat. Cancer 2019, 26, R15–R30. [Google Scholar] [CrossRef] [Green Version]
- Pandey, K.; An, H.-J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int. J. Cancer 2019, 145, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- The, I.; Ruijtenberg, S.; Bouchet, B.P.; Cristobal, A.; Prinsen, M.B.W.; van Mourik, T.; Koreth, J.; Xu, H.; Heck, A.J.R.; Akhmanova, A.; et al. Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat. Commun. 2015, 6, 5906. [Google Scholar] [CrossRef] [Green Version]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol. Cell 2013, 50, 250–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zupkovitz, G.; Grausenburger, R.; Brunmeir, R.; Senese, S.; Tischler, J.; Jurkin, J.; Rembold, M.; Meunier, D.; Egger, G.; Lagger, S.; et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol. Cell. Biol. 2010, 30, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efeyan, A.; Ortega-Molina, A.; Velasco-Miguel, S.; Herranz, D.; Vassilev, L.T.; Serrano, M. Induction of p53-dependent senescence by the MDM2 antagonist nutlin-3a in mouse cells of fibroblast origin. Cancer Res. 2007, 67, 7350–7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef]
- Chen, X.; Xu, D.; Li, X.; Zhang, J.; Xu, W.; Hou, J.; Zhang, W.; Tang, J. Latest Overview of the Cyclin-Dependent Kinases 4/6 Inhibitors in Breast Cancer: The Past, the Present and the Future. J. Cancer 2019, 10, 6608–6617. [Google Scholar] [CrossRef]
- Marra, A.; Curigliano, G. Are all cyclin-dependent kinases 4/6 inhibitors created equal? NPJ Breast Cancer 2019, 5, 27. [Google Scholar] [CrossRef] [Green Version]
- Gervaso, L.; Montero, A.J.; Jia, X.; Khorana, A.A. Venous thromboembolism in breast cancer patients receiving cyclin-dependent kinase inhibitors. J. Thromb. Haemost. 2020, 18, 162–168. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Cheng, C.K.; Gustafson, W.C.; Charron, E.; Houseman, B.T.; Zunder, E.; Goga, A.; Gray, N.S.; Pollok, B.; Oakes, S.A.; James, C.D.; et al. Dual blockade of lipid and cyclin-dependent kinases induces synthetic lethality in malignant glioma. Proc. Natl. Acad. Sci. USA 2012, 109, 12722–12727. [Google Scholar] [CrossRef] [Green Version]
- Matutino, A.; Amaro, C.; Verma, S. CDK4/6 inhibitors in breast cancer: Beyond hormone receptor-positive HER2-negative disease. Adv. Med. Oncol. 2018, 10, 1758835918818346. [Google Scholar] [CrossRef] [Green Version]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2018, 67, 2142–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teh, J.L.F.; Aplin, A.E. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin. Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015-2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Es, K.; Ak, W. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar]
- O’Shaughnessy, J.; Thaddeus Beck, J.; Royce, M. Everolimus-based combination therapies for HR+, HER2- metastatic breast cancer. Cancer Treat. Rev. 2018, 69, 204–214. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
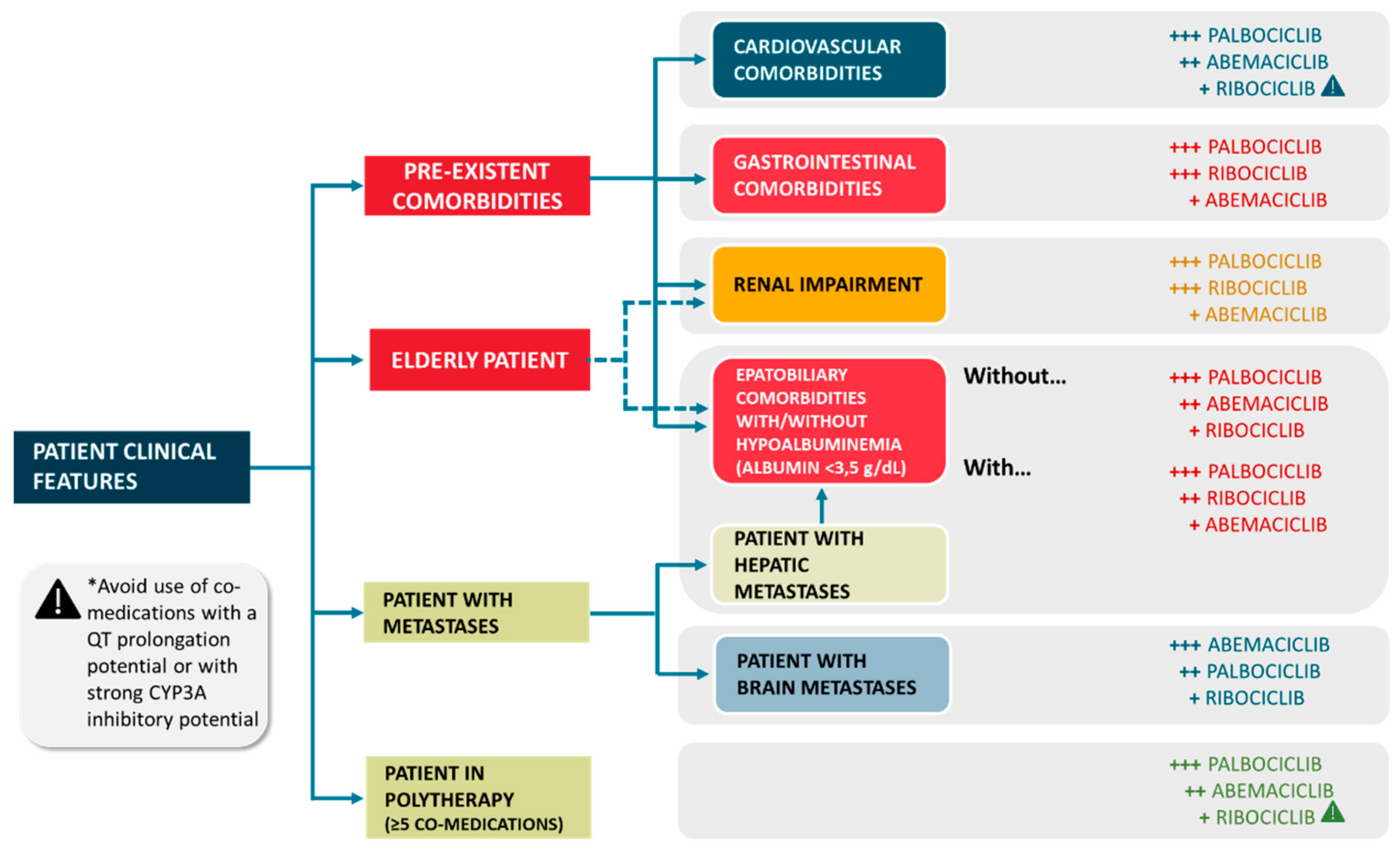{kind=link}
| Targeted Kinase | Pathophysiological Activities of Targeted Kinases | Affinity of Palbociclib | Affinity of Ribociclib | Affinity of Abemaciclib |
|---|---|---|---|---|
| CDK1 | It is mainly involved in controlling the transition from G2 to M phase of cell-cycle | - | - | + |
| CDK2 | It selectively orchestrates processes of phase S, binding Cyclin E, and not Cyclin D as for the other CDKs | - | - | + |
| CDK4 | It inhibits members of the retinoblastoma (RB) protein family including RB1 and regulate the cell-cycle during G(1)/S transition [34] | ++ | +++ | +++ |
| CDK6 | It inhibits members of the retinoblastoma (RB) protein family including RB1 and regulate the cell-cycle during G(1)/S transition [34] | ++ | ++ | + |
| CDK7 | It regulates the initiation of transcription through phosphorylation of the heptad repeats that comprise the C-terminal tail of RNA polymerase II (CTD) | - | - | + |
| CDK9 | It regulates the release from promoter proximal arrest of transcription through phosphorylation of the heptad repeats that comprise the C-terminal tail of RNA polymerase II (CTD) | - | - | ++ |
| GSK3 α/β | It promotes the synthesis of pro-inflammatory IL-6 and the expression of oncogenic genes | - | - | + |
| CAMKII α/β/γ | It is involved in apoptosis and autophagy in cancer cells | - | - | + |
| DYRK | It regulates some proteins controlling the cell cycle | - | - | + |
| PIM protein kinase | It is an oncogenic protein which is frequently amplified in cancer | - | - | + |
| HIPK | It promotes JAK/STAT signaling | - | - | + |
| CAMK families | They are enzymes overexpressed in several cancer types | - | - | + |
| CDKi | Registration Trial | ET Backbone | Patients Reporting Adverse Events with Grade 3 or 4 of (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Neutropenia | Leukopenia | Anemia | Infections | Nausea | Vomiting | Diarrhea | Fatigue | |||
| Palbociclib | PALOMA-2 | AI | 66.5 | 24.8 | 5.4 | 0 | 0.2 | 0.5 | 1.4 | 1.8 |
| PALOMA-3 | Fulvestrant | 76.1 | 33.8 | 2.8 | 3.2 | 0 | 0 | 0 | 0 | |
| Ribociclib | MONALEESA-2 | Letrozole | 59.3 | 21 | 1.2 | 4.2 | 2.4 | 3.6 | 1 | 2.4 |
| MONALEESA-3 | Fulvestrant | 57.1 | 15.5 | 3.9 | 7.7 | 1.4 | 14.1 | 0.6 | 1.7 | |
| Abemaciclib | MONARCH-1 (monotherapy) | - | 28.9 | 27.7 | 0 | - | 4.5 | 1.5 | 19.7 | 12.9 |
| MONARCH-2 | Fulvestrant | 26.5 | 7.3 | 5.8 | 4.9 | 0.9 | 1.2 | 13.4 | 1.8 | |
| MONARCH-3 | AI | 23.8 | 8.6 | 7 | - | 1.2 | 1.5 | 9.5 | 1.8 | |
| Pharmacological Features | Palbociclib [45] | Ribociclib [10] | Abemaciclib [46] | |
|---|---|---|---|---|
| Dosage and schedule | 125 mg/daily day 1–21 Q28 with food | 600 mg/daily day 1–21 Q28 | 200 mg twice daily in monotherapy; 150 mg twice daily in combination with endocrine therapy | |
| Selectivity | CDK4 = CDK6 [27] | CDK4 > CDK6 [28] | CDK4 >> CDK6; low potency to CDK1, CDK7 and CDK9 [31] | |
| Lipophilicity; BBB penetration | cLogP value of 5,5; + [47] | N.A. | cLogP value of 2.7; +++ [47] | |
| PK | Cmax: 52 ng/mL Tmax: 7 h t1/2: 25.9 h Vd: 2793 L AUC 0–10 (ng/mlxh): 299 [48] | Cmax: 1000 ng/mL (higher value for Asiatic people) Tmax: 5 h t1/2:32.6 h Vd: 1090 L AUC 0–24 (ng/mlxh): 20000 [24,49] | Cmax: 298 ng/mL Tmax: 8 h t1/2: 8 h Vd: 690.3 L AUC 0–24 (ng/mlxh): 5520 | |
| Bioavailability | 46% | N.A. | 45% | |
| Binding protein | 85% | 70% | 96–98% | |
| Metabolism | Hepatic: substrate of CYP3A and SULT2A1 [48] | Hepatic: substrate of CYP3A4 | Hepatic: substrate of CYP3A4 | |
| Excretion | In feces | 74% | 69.1% | 81% |
| In urine | 17% | 22.6% | 3.4% | |
| Effect on ADME enzymes + autoinhibition | Weak and time-dependent inhibitor of CYP3A. Palbociclib is a substrate of P-gp and BCRP and inhibits OCT1. [7] | Moderate/strong dose- and time-dependent inhibitor of CYP3A4. Ribociclib is a substrate of P-gp. Reversible CYP1A2, 2E1 inhibitor. Potentially inhibits P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, BSEP and MATE1. | Abemaciclib is a substrate of P-gp and BCRP and inhibits OCT2 and MATE | |
| Active metabolites | No [40] | No | Yes: N-desethylabemaciclib (M2), hydroxyabemaciclib (M20), hydroxy-N-desethylabemaciclib (M18) | |
| Food intake alteration | Absorption and drug exposure lower in fasted state [40] | No | High fat and high caloric meal increase AUC (9%) and Cmax (26%) | |
| Adverse events | Neutropenia G3/4 | Nausea any grade | Diarrhea any grade; Fatigue any grade. Neutropenia (rare and manageable) | |
| Effect of co-administered CYP3A inhibitors | ↑87% AUC ↑34% Cmax | Strong inhibitors: ↑3.2-fold AUC and ↑1.7-fold Cmax; Moderate inhibitors: ↑1.9-fold AUC and ↑1.3-fold Cmax (after a single 400 mg dose) | Strong inhibitors: ↑237% AUC (↑119% of the active metabolites) and ↑30% Cmax (↑7% of the active metabolites)Moderate inhibitors: ↑1.7-fold AUC (↑1,3-fold of the active metabolites) | |
| Effect of co-administered CYP3A inducers | Strong inducers: ↓85% AUC and ↓70% Cmax; Moderate inducers: ↓32% AUC and ↓11% Cmax [7] | Strong inducers: ↓89% AUC ↓81% Cmax Moderate inducers: ↓60%AUC ↓37% Cmax (after a single 600 mg dose) | Strong inducers: ↓67% AUC of parent drug and active metabolites Moderate inducers: not known | |
| Pediatric use | No data | No data | No data | |
| Geriatric use | No differences on safety and efficacy | No differences on safety and efficacy | No differences on safety and efficacy | |
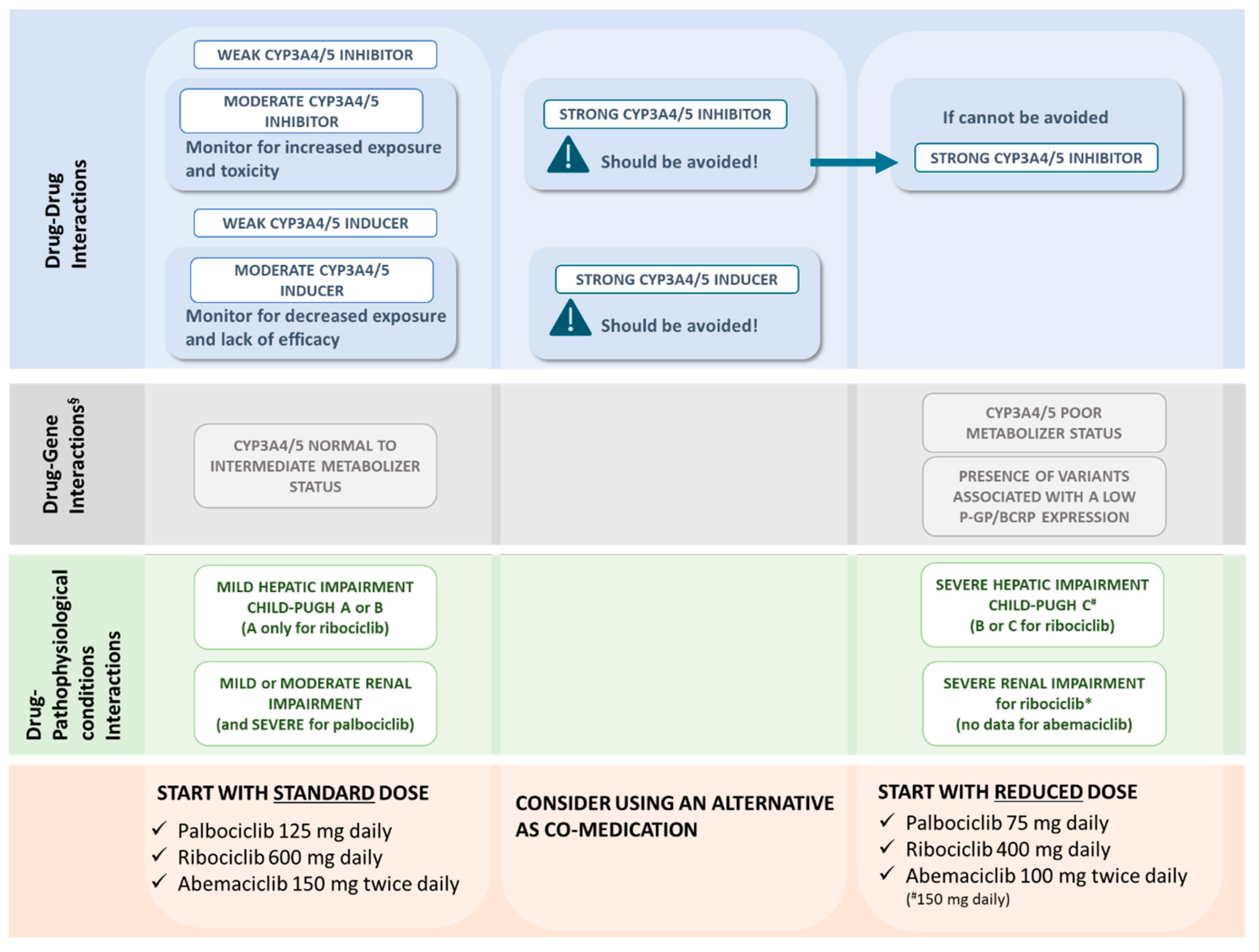
| Concomitant Medications or Pathophysiological Conditions | Palbociclib 125 mg Once a Day, Day 1–21 Q28 | Ribociclib 600 mg Once a Day, Day 1–21 Q28 | Abemaciclib 150 mg Twice Daily in Combination with Endocrine Therapy; 200 mg Twice Daily in Monotherapy. Continuous Schedule |
|---|---|---|---|
| Strong CYP3A inhibitor | Avoid. If unavoidable: 75 mg/day starting dose (↓40%) * | Avoid. If unavoidable: 400 mg/day starting dose (↓33.33%) * | Avoid. If unavoidable: 100 mg twice daily starting dose (↓50/33.33%) * |
| Moderate CYP3A inhibitor | Monitoring | Monitoring | Monitoring |
| Weak CYP3A inhibitor | Low risk of DDI | Low risk of DDI | Low risk of DDI |
| Strong CYP3A inducer | Avoid. Consider an alternative | Avoid. Consider an alternative | Avoid. Consider an alternative |
| Moderate CYP3A inducer | Monitoring | Monitoring | Monitoring |
| Weak CYP3A inducer | Low risk of DDI | Low risk of DDI | Low risk of DDI |
| Hepatic impairment recommendation | Child-Pugh A or B: no modifications Child-Pugh C: 75 mg/day starting dose | Child-Pugh A: no modifications Child-Pugh B or C: 400 mg/day starting dose | Child-Pugh A or B: no modifications Child Pugh C: 150 mg/day starting dose |
| Renal impairment recommendation | Mild to moderate: no modifications Severe or hemodialysis: no data | Mild to moderate: no modifications Severe: lower starting dose to 400 mg/day (EMA) or 200 mg/day starting dose (FDA) | Mild to moderate: no modifications Severe or hemodialysis: no data |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roncato, R.; Angelini, J.; Pani, A.; Cecchin, E.; Sartore-Bianchi, A.; Siena, S.; De Mattia, E.; Scaglione, F.; Toffoli, G. CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene, and Pathophysiological Conditions. Int. J. Mol. Sci. 2020, 21, 6350. https://doi.org/10.3390/ijms21176350
Roncato R, Angelini J, Pani A, Cecchin E, Sartore-Bianchi A, Siena S, De Mattia E, Scaglione F, Toffoli G. CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene, and Pathophysiological Conditions. International Journal of Molecular Sciences. 2020; 21(17):6350. https://doi.org/10.3390/ijms21176350
Chicago/Turabian StyleRoncato, Rossana, Jacopo Angelini, Arianna Pani, Erika Cecchin, Andrea Sartore-Bianchi, Salvatore Siena, Elena De Mattia, Francesco Scaglione, and Giuseppe Toffoli. 2020. "CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene, and Pathophysiological Conditions" International Journal of Molecular Sciences 21, no. 17: 6350. https://doi.org/10.3390/ijms21176350
APA StyleRoncato, R., Angelini, J., Pani, A., Cecchin, E., Sartore-Bianchi, A., Siena, S., De Mattia, E., Scaglione, F., & Toffoli, G. (2020). CDK4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene, and Pathophysiological Conditions. International Journal of Molecular Sciences, 21(17), 6350. https://doi.org/10.3390/ijms21176350