Proteostasis and Proteotoxicity in the Network Medicine Era




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
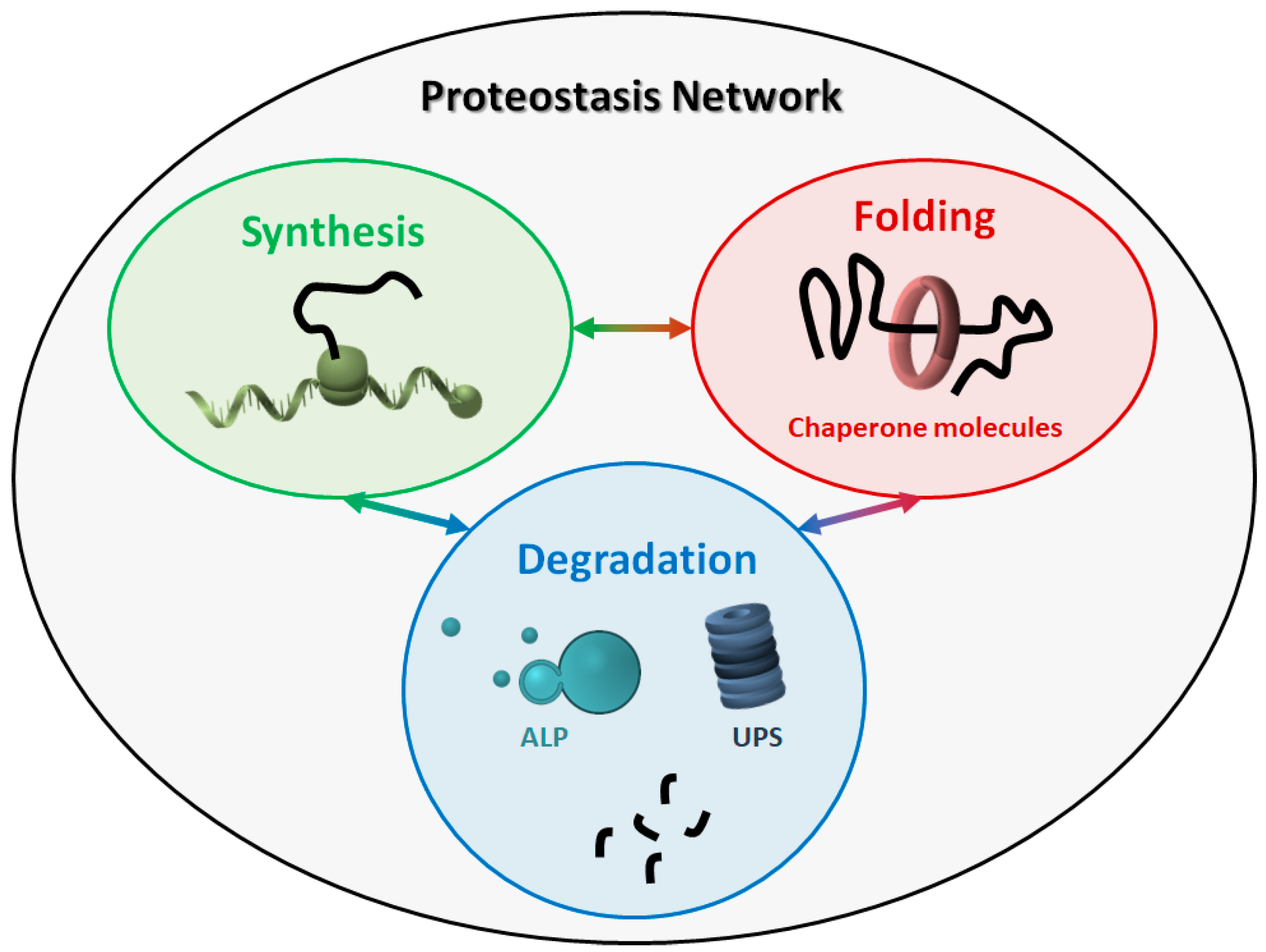
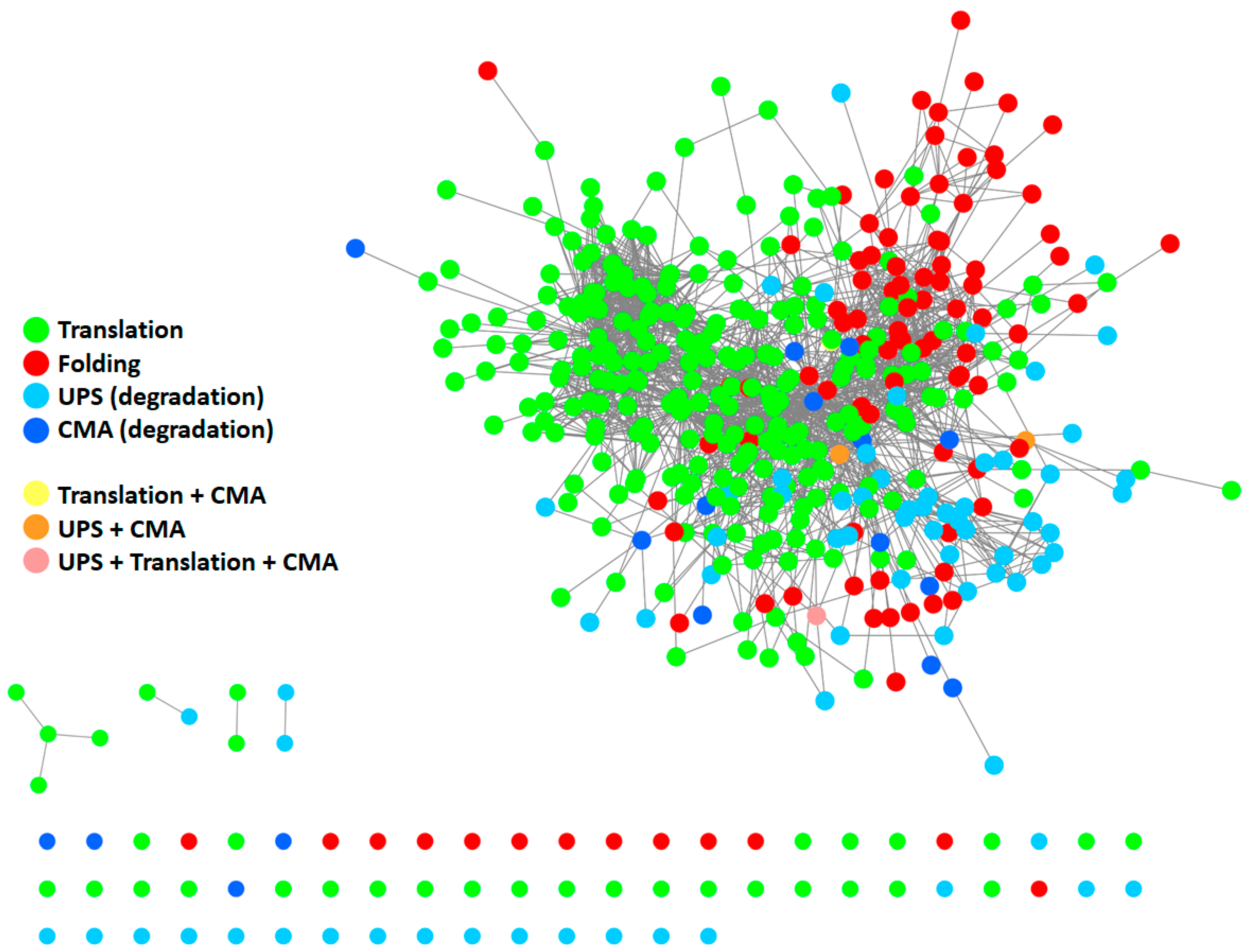
2. The Proteostasis Network
2.1. Molecular Chaperones
2.2. Protein Degradation
3. The Failure of the PN in Neurodegeneration
4. Targeting the PN in Neurodegeneration: “Classical” Pharmacological Approaches
5. Novel Strategies to Ameliorate Proteinopathies: The “Network Medicine” Approach
5.1. Complex Systems and Diseases
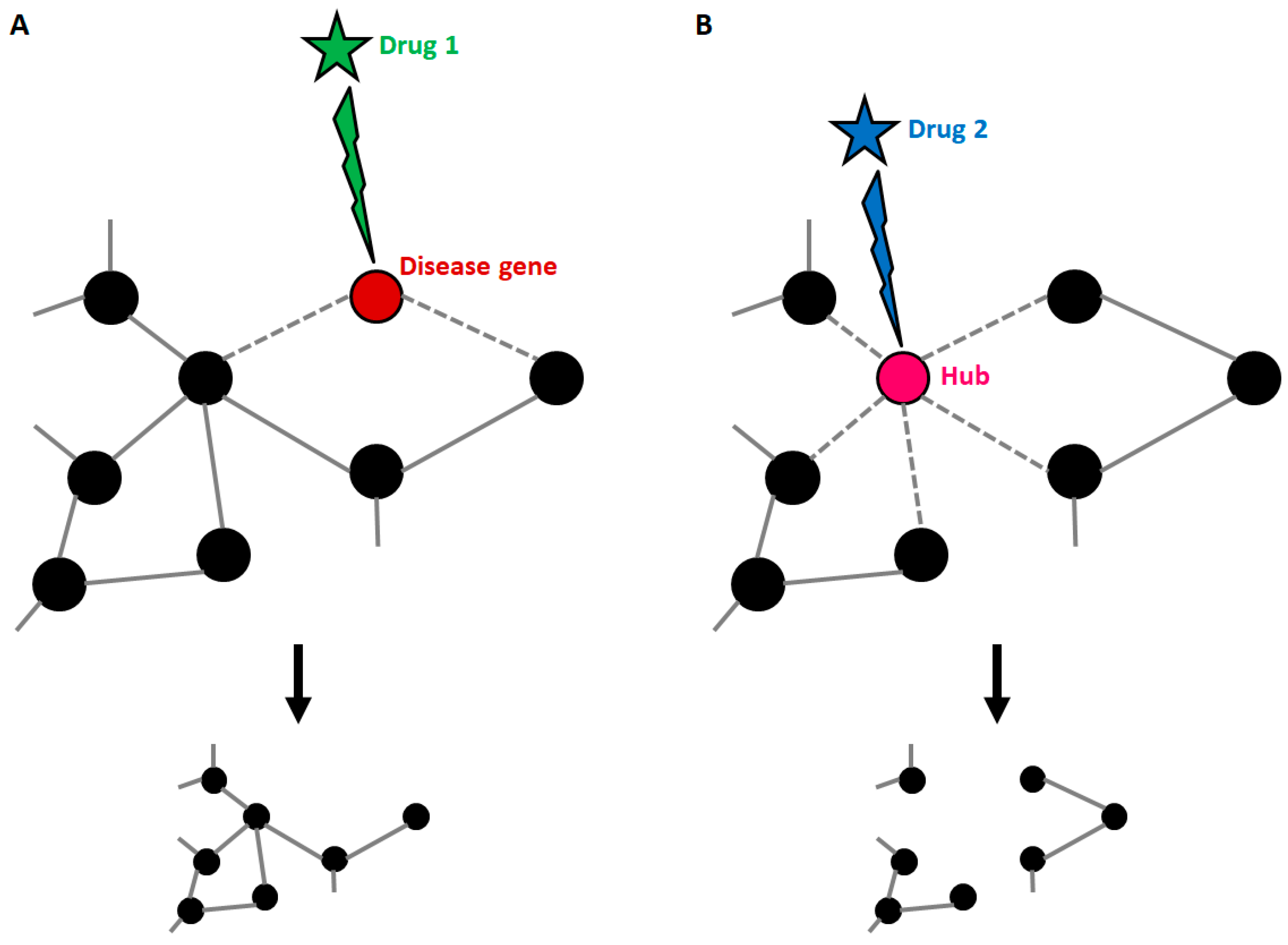
5.2. Network Medicine
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, R.J. Roles of molecular chaperones in protein folding. Curr. Opin. Struct. Biol. 1994, 4, 117–122. [Google Scholar] [CrossRef]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.B.; Dichgans, J. Molecular pathogenesis of movement disorders: Are protein aggregates a common link in neuronal degeneration? Curr. Opin. Neurol. 1999, 12, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Newton, T.M.; Duce, J.A.; Bayle, E.D. The proteostasis network provides targets for neurodegeneration. Br. J. Pharm. 2019, 176, 3508–3514. [Google Scholar] [CrossRef] [Green Version]
- Batta, A.; Kalra, B.S.; Khirasaria, R. Trends in FDA drug approvals over last 2 decades: An observational study. J. Fam. Med. Prim. Care 2020, 9, 105–114. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Mathur, S.; Sutton, J. Personalized medicine could transform healthcare. Biomed. Rep. 2017, 7, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Ginsburg, G.S.; Phillips, K.A. Precision medicine: From science to value. Health Aff. Proj. Hope 2018, 37, 694–701. [Google Scholar] [CrossRef]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays News Rev. Mol. Cell. Dev. Biol. 2013, 35, 1050–1055. [Google Scholar] [CrossRef] [Green Version]
- Zaher, H.S.; Green, R. Fidelity at the molecular level: Lessons from protein synthesis. Cell 2009, 136, 746–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isken, O.; Maquat, L.E. Quality control of eukaryotic mRNA: Safeguarding cells from abnormal mRNA function. Genes Dev. 2007, 21, 1833–1856. [Google Scholar] [CrossRef] [Green Version]
- Pechmann, S.; Willmund, F.; Frydman, J. The ribosome as a hub for protein quality control. Mol. Cell 2013, 49, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedialkova, D.D.; Leidel, S.A. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 2015, 161, 1606–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [Green Version]
- Orchard, S.; Kerrien, S.; Abbani, S.; Aranda, B.; Bhate, J.; Bidwell, S.; Bridge, A.; Briganti, L.; Brinkman, F.S.L.; Brinkman, F.; et al. Protein interaction data curation: The international molecular exchange (IMEx) consortium. Nat. Methods 2012, 9, 345–350. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A chaperome sub-network safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, A. Heat shock proteins: Facts, thoughts, and dreams. Shock 1999, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitley, D.; Goldberg, S.P.; Jordan, W.D. Heat shock proteins: A review of the molecular chaperones. J. Vasc. Surg. 1999, 29, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Rizzolo, K.; Huen, J.; Kumar, A.; Phanse, S.; Vlasblom, J.; Kakihara, Y.; Zeineddine, H.A.; Minic, Z.; Snider, J.; Wang, W.; et al. Features of the chaperone cellular network revealed through systematic interaction mapp. Cell Rep. 2017, 20, 2735–2748. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I. Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Zhang, T.; Shen, S.; Qu, J.; Ghaemmaghami, S. Global analysis of cellular protein flux quantifies the selectivity of basal autophagy. Cell Rep. 2016, 14, 2426–2439. [Google Scholar] [CrossRef] [Green Version]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, A.J.; Bott, L.C.; Morimoto, R.I. Shaping proteostasis at the cellular, tissue, and organismal level. J. Cell Biol. 2017, 216, 1231–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef]
- Yu, A.; Shibata, Y.; Shah, B.; Calamini, B.; Lo, D.C.; Morimoto, R.I. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc. Natl. Acad. Sci. USA 2014, 111, E1481–E1490. [Google Scholar] [CrossRef] [Green Version]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Roth, D.M.; Hutt, D.M.; Tong, J.; Bouchecareilh, M.; Wang, N.; Seeley, T.; Dekkers, J.F.; Beekman, J.M.; Garza, D.; Drew, L.; et al. Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol. 2014, 12, e1001998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisén, S.; Bertelsen, E.B.; Thompson, A.D.; Patury, S.; Ung, P.; Chang, L.; Evans, C.G.; Walter, G.M.; Wipf, P.; Carlson, H.A.; et al. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of Hsp70–Hsp40. ACS Chem. Biol. 2010, 5, 611–622. [Google Scholar] [CrossRef]
- Wang, A.M.; Miyata, Y.; Klinedinst, S.; Peng, H.-M.; Chua, J.P.; Komiyama, T.; Li, X.; Morishima, Y.; Merry, D.E.; Pratt, W.B.; et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol. 2013, 9, 112–118. [Google Scholar] [CrossRef]
- Miyata, Y.; Li, X.; Lee, H.-F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D.; et al. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Sun, W.; Taldone, T.; Rodina, A.; Chiosis, G. Heat shock protein 90 in neurodegenerative diseases. Mol. Neurodegener. 2010, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef] [Green Version]
- Blair, L.J.; Sabbagh, J.J.; Dickey, C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets 2014, 18, 1219–1232. [Google Scholar] [CrossRef] [Green Version]
- Spreafico, A.; Delord, J.-P.; De Mattos-Arruda, L.; Berge, Y.; Rodon, J.; Cottura, E.; Bedard, P.L.; Akimov, M.; Lu, H.; Pain, S.; et al. A first-in-human phase I, dose-escalation, multicentre study of HSP990 administered orally in adult patients with advanced solid malignancies. Br. J. Cancer 2015, 112, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, A.; Kubota, H.; Pack, C.-G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lee, D.; Lee, J.; Song, H.; Kim, H.-J.; Kim, K.-T. Vaccinia-related kinase 2 controls the stability of the eukaryotic chaperonin TRiC/CCT by inhibiting the deubiquitinating enzyme USP25. Mol. Cell Biol. 2015, 35, 1754–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neef, D.W.; Jaeger, A.M.; Thiele, D.J. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat. Rev. Drug Discov. 2011, 10, 930–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neef, D.W.; Turski, M.L.; Thiele, D.J. Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol. 2010, 8, e1000291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neef, D.W.; Jaeger, A.; Gomez-Pastor, R.; Willmund, F.; Frydman, J.; Thiele, D.J. A direct regulatory interaction between chaperonin TRiC and stress responsive transcription factor HSF1. Cell Rep. 2014, 9, 955–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome activation by small molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Serio, A.; Arjun, A.; Bilican, B.; Daub, A.; Ando, D.M.; Tsvetkov, A.; Pleiss, M.; Li, X.; Peisach, D.; et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat. Chem. Biol. 2014, 10, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Kuo, S.-Y.; Castoreno, A.B.; Aldrich, L.N.; Lassen, K.G.; Goel, G.; Dančík, V.; Kuballa, P.; Latorre, I.; Conway, K.L.; Sarkar, S.; et al. Small-molecule enhancers of autophagy modulate cellular disease phenotypes suggested by human genetics. Proc. Natl. Acad. Sci. USA 2015, 112, E4281–E4287. [Google Scholar] [CrossRef] [Green Version]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Ma’ayan, A. Complex systems biology. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lualdi, M.; Fasano, M. Statistical analysis of proteomics data: A review on feature selection. J. Proteom. 2019, 198, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.; Barabási, A.-L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47–97. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J.; Kohane, I.; Barabasi, A.-L. Human disease classification in the postgenomic era: A complex systems approach to human pathobiology. Mol. Syst. Biol. 2007, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Hao, T.; Shaw, C.; Patel, A.J.; Szabó, G.; Rual, J.-F.; Fisk, C.J.; Li, N.; Smolyar, A.; Hill, D.E.; et al. A protein-protein interaction network for human inherited ataxias and disorders of purkinje cell degeneration. Cell 2006, 125, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, M.A.; Goh, K.-I.; Cusick, M.E.; Barabási, A.-L.; Vidal, M. Drug-target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef]
- Lehtonen, Š.; Sonninen, T.-M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of cellular proteostasis in Parkinson’s disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Kovács, I.A.; Barabási, A.-L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lualdi, M.; Alberio, T.; Fasano, M. Proteostasis and Proteotoxicity in the Network Medicine Era. Int. J. Mol. Sci. 2020, 21, 6405. https://doi.org/10.3390/ijms21176405
Lualdi M, Alberio T, Fasano M. Proteostasis and Proteotoxicity in the Network Medicine Era. International Journal of Molecular Sciences. 2020; 21(17):6405. https://doi.org/10.3390/ijms21176405
Chicago/Turabian StyleLualdi, Marta, Tiziana Alberio, and Mauro Fasano. 2020. "Proteostasis and Proteotoxicity in the Network Medicine Era" International Journal of Molecular Sciences 21, no. 17: 6405. https://doi.org/10.3390/ijms21176405
APA StyleLualdi, M., Alberio, T., & Fasano, M. (2020). Proteostasis and Proteotoxicity in the Network Medicine Era. International Journal of Molecular Sciences, 21(17), 6405. https://doi.org/10.3390/ijms21176405