Combination of Linkage Mapping, GWAS, and GP to Dissect the Genetic Basis of Common Rust Resistance in Tropical Maize Germplasm

 , , ,
, , ,  , , and
, , and 
Abstract
:1. Introduction
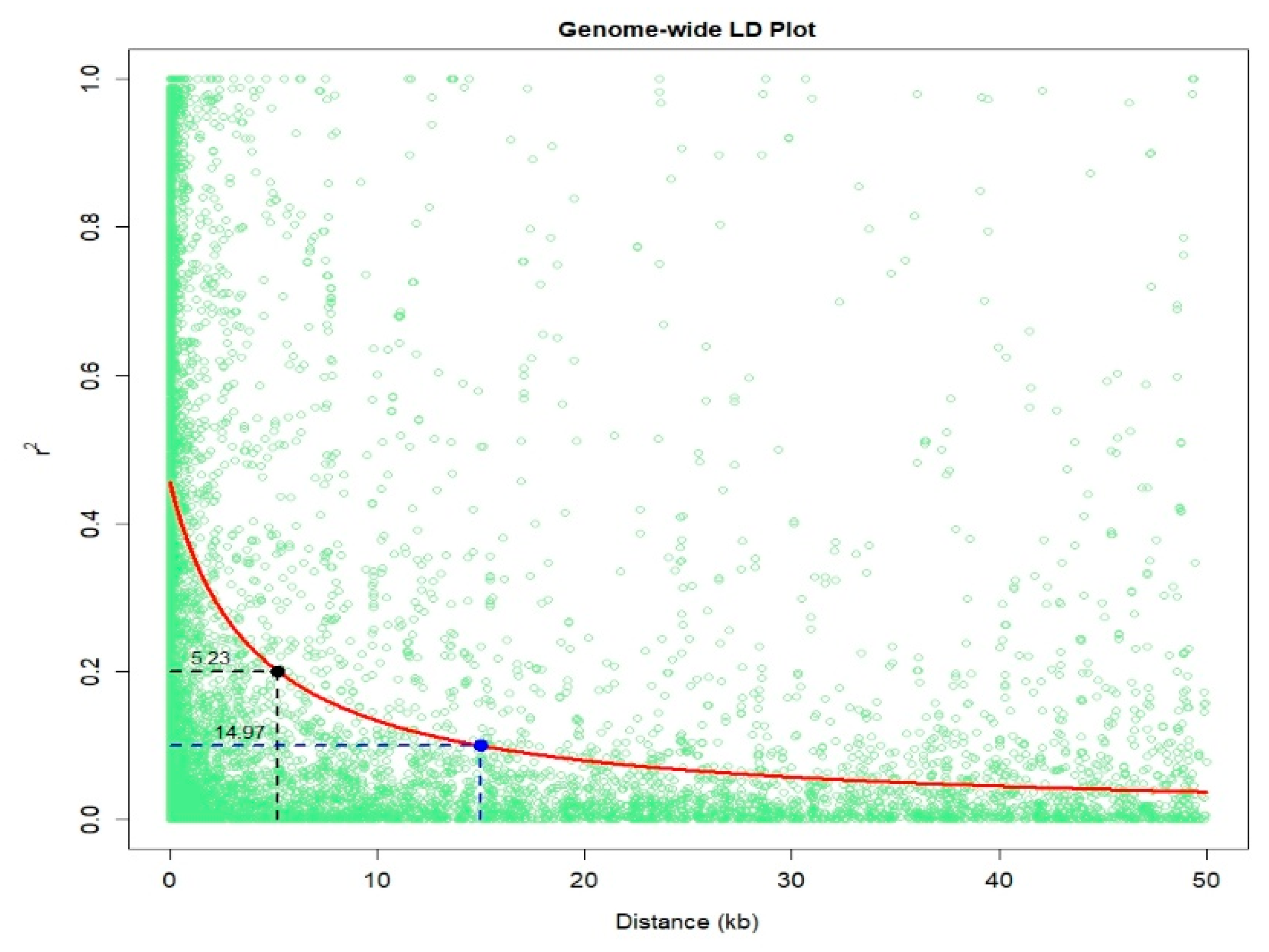
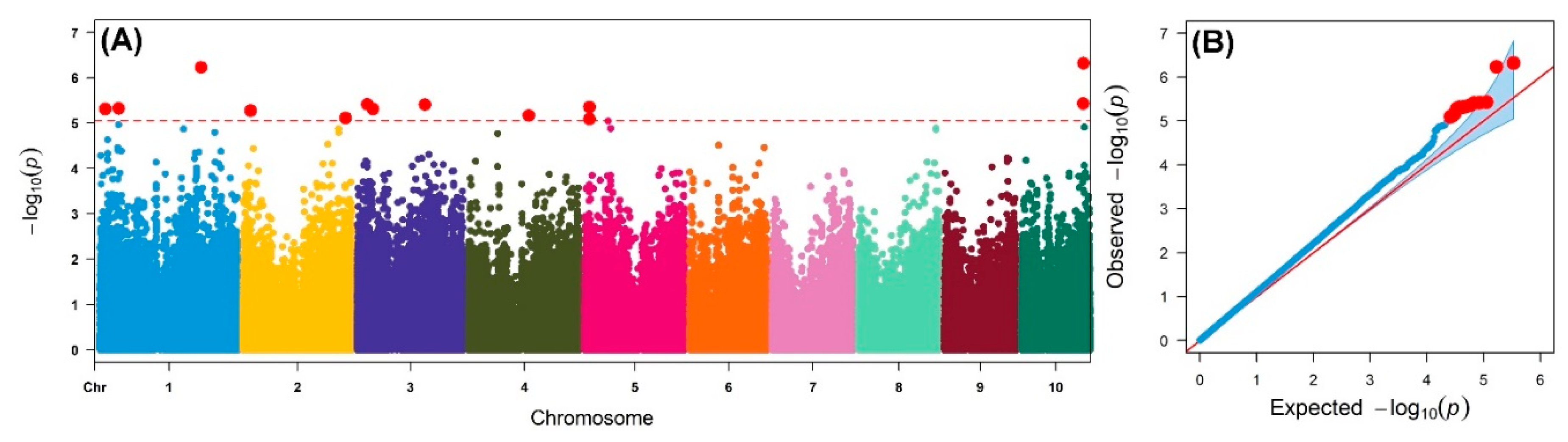
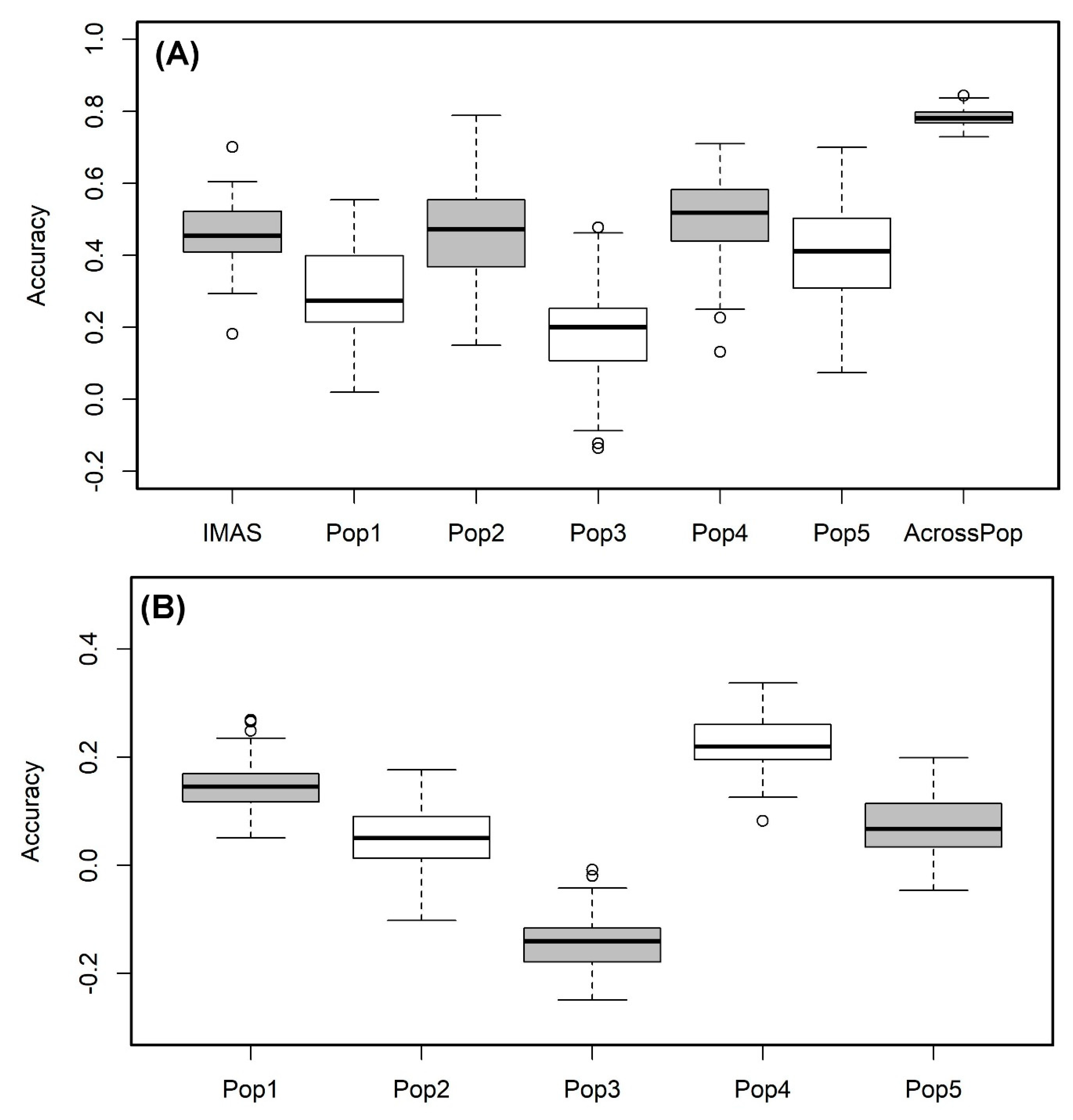
2. Results
3. Discussion
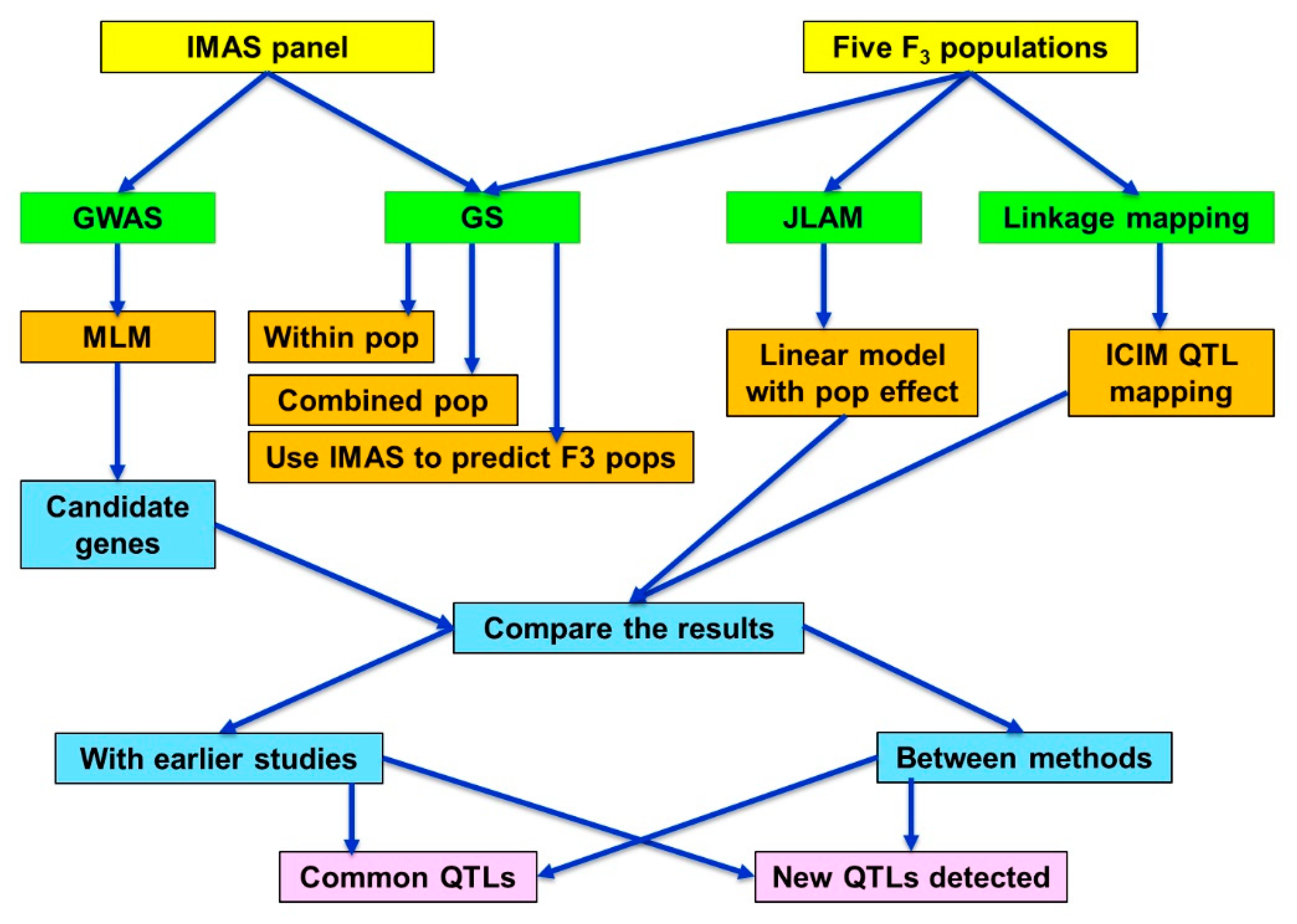
4. Materials and Methods
4.1. Plant Materials and Trial Design
4.2. Phenotypic and Genotypic Data Analysis
Block(Rep.Env)pno + emnop
4.3. PCA and Linkage Disequilibrium
4.4. Genome-wide Association Analyses
4.5. Detection of QTLs and Joint Linkage Association Mapping
4.6. Genomic Prediction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dey, U.; Harlapur, S.I.; Dhutraj, D.N.; Suryawanshi, A.P.; Bhattacharjee, R. Integrated disease management strategy of common rust of maize incited by Puccinia sorghi Schw. Afr. J. Microbiol. Res. 2015, 9, 1345–1351. [Google Scholar]
- Dey, U.; Harlapur, S.I.; Dhutraj, D.N.; Suryawanshi, A.P.; Badgujar, S.L.; Jagtap, G.P.; Kuldhar, D.P. Spatiotemporal yield loss assessment in corn due to common rust caused by Puccinia sorghi Schw. Afr. J. Agric. Res. 2012, 7, 5265–5269. [Google Scholar] [CrossRef]
- Crautam, P.; Stein, J. Induction of systemic acquired resistance to Puccinia sorghi in corn. Inter. J. Plant Pathol. 2011, 2, 43–50. [Google Scholar]
- Chandler, M.A.; Tracy, W.F. Vegetative phase change among sweet corn (Zea mays L.) hybrids varying for reaction to common rust (Puccinia sorghi Schw.). Plant Breed. 2007, 126, 569–573. [Google Scholar] [CrossRef]
- Abedon, B.G.; Tracy, W.F. Corngrass 1 of maize (Zea mays L.) delays development of adult plant resistance to common rust (Puccinia sorghi Schw.) and European corn borer (Ostrinia nubilalis Hubner). J. Hered. 1996, 87, 219–223. [Google Scholar] [CrossRef]
- Hurkman, M.M. Vegetative Phase Transition and Response to Disease in Maize (Zea mays L.). Ph.D. Thesis, University of Wisconsin, Madison, WI, USA, 2003. Available online: https://elibrary.ru/item.asp?id=5282527 (accessed on 15 August 2020).
- Danson, J.; Lagat, M.; Kimani, M.; Kuria, A. Quantitative trait loci (QTLs) for resistance to gray leaf spot and common rust diseases of maize. African, J. Biotechnol. 2008, 7, 3247–3254. [Google Scholar]
- Ayiga-Aluba, J.; Edemal, R.; Tusiime, G.; Asea, G.; Gibson, P. Response to two cycles of S1 recurrent selection for turcicum leave blight in an open pollinated maize variety population (Longe 5). Adv. Appl. Sci. Res. 2015, 6, 4–12. [Google Scholar]
- Delaney, D.E.; Webb, C.A.; Hulbert, S.H. A novel rust resistance gene in maize showing overdominance. Mol. plant-microbe Interact. 1998, 11, 242–245. [Google Scholar] [CrossRef]
- Lübberstedt, T.; Klein, D.; Melchinger, A.E. Comparative quantitative trait loci mapping of partial resistance to Puccinia sorghi across four populations of European flint maize. Phytopathology 1998, 88, 1324–1329. [Google Scholar] [CrossRef] [Green Version]
- Hooker, A.L. Corn and sorghum rusts. In Diseases, Distribution, Epidemiology, and Control; Elsevier: Amsterdam, The Netherlands, 1985; pp. 207–236. [Google Scholar]
- Olukolu, B.A.; Tracy, W.F.; Wisser, R.; De Vries, B.; Balint-Kurti, P.J. A Genome-Wide Association Study for Partial Resistance to Maize Common Rust. Phytopathology 2016, 106, 745–751. [Google Scholar] [CrossRef]
- Brown, A.F.; Juvik, J.A.; Pataky, J.K. Quantitative trait loci in sweet corn associated with partial resistance to Stewart’s wilt, northern corn leaf blight, and common rust. Phytopathology 2001, 91, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; McMullen, M.D.; Ware, D. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163. [Google Scholar] [CrossRef] [PubMed]
- Zila, C.T.; Fernando Samayoa, L.; Santiago, R.; Butrón, A.; Holland, J.B. A Genome-Wide Association Study Reveals Genes Associated with Fusarium Ear Rot Resistance in a Maize Core Diversity Panel. G3 (Bethesda) 2013, 3, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laidò, G.; Marone, D.; Russo, M.A.; Colecchia, S.A.; Mastrangelo, A.M.; De Vita, P.; Papa, R. Linkage Disequilibrium and Genome-Wide Association Mapping in Tetraploid Wheat (Triticum turgidum L.). PLoS ONE 2014, 9, e95211. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Ali, F.; Chen, G.; Li, H.; Mahuku, G.; Yang, N.; Narro, L.; Magorokosho, C.; Makumbi, D.; Yan, J. Genome-wide association mapping reveals novel sources of resistance to northern corn leaf blight in maize. BMC Plant Biol. 2015, 15, 206. [Google Scholar] [CrossRef] [Green Version]
- Gowda, M.; Das, B.; Makumbi, D.; Babu, R.; Semagn, K.; Mahuku, G.; Olsen, M.S.; Bright, J.M.; Beyene, Y.; Prasanna, B.M. Genome-wide association and genomic prediction of resistance to maize lethal necrosis disease in tropical maize germplasm. Theor. Appl. Genet. 2015, 128, 1957–1968. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Luo, J.; Qi, C.; Ruan, Y.; Li, J.; Zhang, A.; Yang, X.; He, Y. Genome-wide association study (GWAS) reveals the genetic architecture of four husk traits in maize. BMC Genomics 2016, 17, 946. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, T.J.; de Leon, N.; Kaeppler, S.M.; Tracy, W.F. Genetic Analysis of Sugarcane mosaic virus Resistance in the Wisconsin Diversity Panel of Maize. Crop Sci. 2018, 58, 1853–1865. [Google Scholar] [CrossRef]
- Liu, N.; Xue, Y.; Guo, Z.; Li, W.; Tang, J. Genome-Wide Association Study Identifies Candidate Genes for Starch Content Regulation in Maize Kernels. Front. Plant Sci. 2016, 7, 1046. [Google Scholar] [CrossRef]
- Kuki, M.C.; Scapim, C.A.; Rossi, E.S.; Mangolin, C.A.; do Amaral Junior, A.T.; Pinto, R.J.B. Genome wide association study for gray leaf spot resistance in tropical maize core. PLoS ONE 2018, 13, e0199539. [Google Scholar] [CrossRef]
- Chaikam, V.; Gowda, M.; Nair, S.; Melchinger, A.; Prasanna, B. Genome-wide association study to identify genomic regions influencing spontaneous fertility in maize haploids. Euphytica 2019, 215, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyaga, C.; Gowda, M.; Beyene, Y.; Muriithi, W.; Makumbi, D.; Olsen, M.; Suresh, L.M.; Bright, J.; Das, B.; Prasanna, B. Genome-Wide Analyses and Prediction of Resistance to MLN in Large Tropical Maize Germplasm. Genes 2019, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, V.; Gupta, S.; Gahlaut, V.; Muthamilarasan, M.; Bandyopadhyay, T.; Ramchiary, N.; Prasad, M. Genome-Wide Association Study of Major Agronomic Traits in Foxtail Millet (Setaria italica L.) Using ddRAD Sequencing. Sci. Rep. 2019, 9, 5020. [Google Scholar] [CrossRef] [PubMed]
- Sitonik, C.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Makumbi, D.; Oliver, K.; Das, B.; Bright, J.M.; Mugo, S.; Crossa, J.; et al. Genetic architecture of maize chlorotic mottle virus and maize lethal necrosis through GWAS, linkage analysis and genomic prediction in tropical maize germplasm. Theor. Appl. Genet. 2019, 132, 2381–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myles, S.; Peiffer, J.; Brown, P.J.; Ersoz, E.S.; Zhang, Z.; Costich, D.E.; Buckler, E.S. Association mapping: Critical considerations shift from genotyping to experimental design. Plant Cell 2009, 21, 2194–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, J.B. Genetic architecture of complex traits in plants. Curr. Opin. Plant Biol. 2007, 10, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [Green Version]
- Kerns, M.R.; Dudley, J.W.; Rufener, G.K. QTL for resistance to common rust and smut in maize. Maydica 1999, 44, 37–45. [Google Scholar]
- Zheng, H.; Chen, J.; Mu, C.; Makumbi, D.; Xu, Y.; Mahuku, G. Combined linkage and association mapping reveal QTL for host plant resistance to common rust (Puccinia sorghi) in tropical maize. BMC Plant Biol. 2018, 18, 310. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Chao, S.; Asoro, F.G.; Heffner, E.L.; Hayashi, T.; Iwata, H.; Smith, K.P.; Sorrells, M.E.; Jannink, J.L. Genomic Selection in Plant Breeding: Knowledge and Prospects. Adv. Agron. 2011, 110, 77–123. [Google Scholar]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Massman, J.M.; Gordillo, A.; Lorenzana, R.E.; Bernardo, R. Genomewide predictions from maize single-cross data. Theor. Appl. Genet. 2013, 126, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ertiro, B.T.; Labuschagne, M.; Olsen, M.; Das, B.; Prasanna, B.M.; Gowda, M. Genetic dissection of nitrogen use efficiency in tropical maize through genome-wide association and genomic prediction. Front. Plant Sci. 2020, 11, 474. [Google Scholar] [CrossRef] [PubMed]
- Kaler, A.; Gillman, J.; Beissinger, T.; Purcell, L. Comparing Different Statistical Models and Multiple Testing Corrections for Association Mapping in Soybean and Maize. Front. Plant Sci. 2020, 10, 1794. [Google Scholar] [CrossRef]
- Wisser, R.J.; Balint-Kurti, P.J.; Nelson, R.J. The genetic architecture of disease resistance in maize: A synthesis of published studies. Phytopathology 2006, 96, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zeng, B.; Zhao, H.; Zhang, M.; Xie, S.; Lai, J. Genome-wide Transcription Factor Gene Prediction and their Expressional Tissue-Specificities in Maize, F.J. Integr. Plant Biol. 2012, 54, 616–630. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, X.; He, K.; Liu, M.; Li, J.; Gao, Z.; Lin, Z.; Zhang, Y.; Wang, X.; Qiu, X.; et al. The MYB Transcription Factor Superfamily of Arabidopsis: Expression Analysis and Phylogenetic Comparison with the Rice MYB Family. Plant Mol. Biol. 2006, 60, 107–124. [Google Scholar]
- Chauhan, J.S.; Mishra, N.K.; Raghava, G.P.S. Identification of ATP binding residues of a protein from its primary sequence. BMC Bioinformatics 2009, 10, 434. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.S.; Shaw, R.; Winkelmann, J.C.; Shaw, G. Binding of PH domains of β-adrenergic-receptor kinase and β-spectrin to WD40/β-transducin repeat containing regions of the β-subunit of trimeric G-proteins. Biochem. Biophys. Res. Commun. 1994, 203, 29–35. [Google Scholar] [CrossRef]
- Stone, J.M.; Walker, J.C. Plant protein kinase families and signal transduction. Plant Physiol. 1995, 108, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Nashev, L.G.; Chandsawangbhuwana, C.; Balazs, Z.; Atanasov, A.G.; Dick, B.; Frey, F.J.; Baker, M.E.; Odermatt, A. Hexose-6-phosphate Dehydrogenase Modulates 11β-Hydroxysteroid Dehydrogenase Type 1-Dependent Metabolism of 7-keto- and 7β-hydroxy-neurosteroids. PLoS ONE 2007, 2, e561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadov, J.; Sun, X.; Gao, Y.; Ochsenfeld, C.; Bakker, E.; Ren, R.; Flora, J.; Wang, X.; Kumpatla, S.; Meyer, D. Combining powers of linkage and association mapping for precise dissection of QTL controlling resistance to gray leaf spot disease in maize (Zea mays L.). BMC Genomics 2015, 16, 916. [Google Scholar] [CrossRef] [Green Version]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Lorenz, A.J.; Smith, K.P.; Jannink, J. Potential and optimization of genomic selection for Fusarium head blight resistance in six-row barley. Crop Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Rutkoski, J.E.; Poland, J.A.; Singh, R.P.; Huerta-Espino, J.; Bhavani, S.; Barbier, H.; Rouse, M.N.; Jannink, J.; Sorrells, M.E. Genomic selection for quantitative adult plant stem rust resistance in wheat. Plant Genome 2014, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rutkoski, J.; Benson, J.; Jia, Y.; Brown-Guedira, G.; Jannink, J.; Sorrells, M. Evaluation of genomic prediction methods for Fusarium head blight resistance in wheat. Plant Genome 2012, 5, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.; Rutkoski, J. Advances and challenges in genomic selection for disease resistance. Annu. Rev. Phytopathol. 2016, 54, 79–98. [Google Scholar] [CrossRef]
- Beyene, Y.; Gowda, M.; Olsen, M.; Robbins, K.R.; Pérez-Rodríguez, P.; Alvarado, G.; Dreher, K.; Gao, S.Y.; Mugo, S.; Prasanna, B.M.; et al. Empirical Comparison of Tropical Maize Hybrids Selected Through Genomic and Phenotypic Selections. Front. Plant Sci. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- de Roos, A.P.W.; Hayes, B.J.; Goddard, M.E. Reliability of genomic predictions across multiple populations. Genetics 2009, 183, 1545–1553. [Google Scholar] [CrossRef] [Green Version]
- Semagn, K.; Beyene, Y.; Warburton, M.; Tarekegne, A.; Mugo, S.; Meisel, B.; Sehabiague, P.; Prasanna, B.M. Meta-analyses of QTL for grain yield and anthesis silking interval in 18 maize populations evaluated under water-stressed and well-watered environments. BMC Genomics. 2013, 14, 313. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pérez-Rodríguez, P.; Semagn, K.; Beyene, Y.; Babu, R.; López-Cruz, M.A.; Vicente, F.S.; Olsen, M.; Buckler, E.; Jannink, J.-L.; et al. Genomic prediction in biparental tropical maize populations in water-stressed and well-watered environments using low-density and GBS SNPs. Heredity (Edinb) 2015, 114, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmour, A.R.; Gogel, B.J.; Cullis, B.R.; Thompson, R. ASReml User Guide Release 3.0; VSN International Ltd.: Hemel Hempstead, UK, 2009. [Google Scholar]
- Alvarado, G.; López, M.; Vargas, M.; Pacheco, Á.; Rodríguez, F.; Burgueño, J.; Crossa, J. META-R (Multi Environment Trail Analysis with R for Windows)-CIMMYT Research Data & Software Repository Network. (Version 5.0). Available online: https://hdl.handle.net/11529/10201 (accessed on 14 August 2020).
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, P.; Zhang, Z.; Kroon, D.E.; Casstevens, T.; Ramdoss, Y.; Buckler, E. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988, 33, 54–78. [Google Scholar] [CrossRef]
- Meng, L.; Li, H.; Zhang, L.; Wang, J. QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop, J. 2015, 3, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Würschum, T.; Liu, W.; Gowda, M.; Maurer, H.P.; Fischer, S.; Schechert, A.; Reif, J.C. Comparison of biometrical models for joint linkage association mapping. Heredity (Edinb). 2012, 108, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Gowda, M.; Steinhoff, J.; Maurer, H.P.; Würschum, T.; Longin, C.F.H.; Cossic, F.; Reif, J.C. Association mapping in an elite maize breeding population. Theor. Appl. Genet. 2011, 123, 847–858. [Google Scholar] [CrossRef]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- SAS. SAS, Version 9.4; Intelligence Platform: System Admin- Istration Guide; SAS Institute Inc.: Cary, NC, USA, 2015. [Google Scholar]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Utz, H.F.; Melchinger, A.E.; Schön, C.C. Bias and sampling error of the estimated proportion of genotypic variance explained by quantitative trait loci determined from experimental data in maize using cross validation and validation with independent samples. Genetics 2000, 154, 1839–1849. [Google Scholar]
- Zhao, Y.; Gowda, M.; Liu, W.; Würschum, T.; Maurer, H.P.; Longin, F.H.; Ranc, N.; Reif, J.C. Accuracy of genomic selection in European maize elite breeding populations. Theor. Appl. Genet. 2012, 124, 769–776. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Mean (Range) | σ2G | σ2GxE | σ2e | h2 |
|---|---|---|---|---|---|
| IMAS AMP | 3.80 (2.40–5.90) | 0.07 * | 0.03 * | 0.13 | 0.68 |
| CZL0618 × LaPostaSeqC7-F71-1-2-1-1B–Pop1 | 3.61 (2.00–5.50) | 0.024 * | 0.01 | 0.10 | 0.44 |
| CZL074 × LaPostaSeqC7-F103-1-2-1-1B–Pop2 | 3.70 (2.50–5.30) | 0.02 * | 0.02 * | 0.12 | 0.43 |
| CZL00009 × CZL99017–Pop3 | 3.42 (2.32–5.00) | 0.03 * | 0.02 * | 0.11 | 0.45 |
| CML505 × CZL99017–Pop4 | 4.10 (2.76–5.14) | 0.01 * | 0.01 | 0.12 | 0.37 |
| CZL0723 × CZL0724–Pop5 | 4.02 (2.51–6.02) | 0.03 * | 0.02 * | 0.16 | 0.38 |
| Across five populations | 3.85 (2.02–6.00) | 0.04 * | 0.10 * | 0.15 | 0.45 |
| SNP a | Chr | MLM-P Value | R2 (%) | MAF | Allele | Putative Candidate Genes | Predicted Function of Candidate Gene |
|---|---|---|---|---|---|---|---|
| S1_12663024 | 1 | 4.86 × 10−6 | 7.00 | 0.14 | A/G | GRMZM2G480386 | uncharacterized |
| S1_41433126 | 1 | 4.69 × 10−6 | 7.88 | 0.05 | G/A | GRMZM5G886521 | uncharacterized |
| S1_220067760 | 1 | 5.86 × 10−7 | 7.80 | 0.46 | C/T | GRMZM2G564469 | uncharacterized |
| S2_16361185 | 2 | 5.23 × 10−6 | 7.70 | 0.08 | C/T | GRMZM2G086484 | Pleckstrin homology (PH) domain superfamily protein |
| S2_222274747 | 2 | 7.65 × 10−6 | 7.10 | 0.05 | C/T | GRMZM2G009188 | 11-beta-hydroxysteroid dehydrogenase 1B |
| S3_21856582 | 3 | 3.84 × 10−6 | 9.54 | 0.42 | A/C | GRMZM2G395983 | uncharacterized |
| S3_34683394 | 3 | 4.83 × 10−6 | 9.02 | 0.35 | A/G | GRMZM5G881063 | uncharacterized |
| S3_147013779 | 3 | 3.89 × 10−6 | 7.70 | 0.17 | G/C | GRMZM2G060540 | uncharacterized |
| S4_130478096 | 4 | 6.72 × 10−6 | 6.93 | 0.12 | A/T | GRMZM5G833902 | uncharacterized |
| S5_10087070 | 5 | 8.02 × 10−6 | 6.25 | 0.06 | A/G | GRMZM2G181002 | Phosphotransferases, Serine or threonine-specific kinase subfamily |
| S5_10089138 | 5 | 4.39 × 10−6 | 6.56 | 0.07 | T/C | GRMZM2G181002 | |
| S5_51353429 | 5 | 9.05 × 10−6 | 7.40 | 0.19 | G/A | GRMZM2G457211 | uncharacterized |
| S10_134585613 | 10 | 3.68 × 10−6 | 6.65 | 0.12 | C/T | GRMZM2G322582 | ATP binding protein |
| S10_134831452 | 10 | 4.78 × 10−7 | 8.22 | 0.14 | A/G | GRMZM2G181030 | MYB-related transcription factor family that regulates hypocotyl growth by regulating free auxin levels in a time-of-day specific manner (RVE1) |
| QTL Name | Chr | Position (cM) | LOD | PVE (%) | Add | Dom | Total PVE (%) | Left Marker | Right Marker |
|---|---|---|---|---|---|---|---|---|---|
| CZL0618 × LaPostaSeqC7-F71-1-2-1-1B–Pop1 | |||||||||
| qCR1-78 | 1 | 551 | 4.41 | 3.32 | −0.20 | −0.08 | 14.95 | S1_77801418 | S1_80167797 |
| qCR1-290 | 1 | 799 | 2.82 | 3.59 | 0.18 | −0.13 | S1_290957469 | S1_285979058 | |
| qCR2-198 | 2 | 187 | 3.17 | 6.70 | −0.34 | −0.53 | S2_198394488 | S2_230388748 | |
| qCR3-113 | 3 | 307 | 2.85 | 6.50 | 0.26 | −0.24 | S3_224567900 | S3_113425715 | |
| qCR6-38 | 6 | 194 | 2.79 | 3.62 | 0.21 | −0.01 | S6_37902339 | S6_63537451 | |
| qCR6-63 | 6 | 197 | 4.51 | 3.27 | −0.21 | −0.02 | S6_63537451 | S6_65299800 | |
| qCR6-146 | 6 | 446 | 3.09 | 7.03 | 0.30 | −0.42 | S6_147225115 | S6_146382028 | |
| CZL074 × LaPostaSeqC7-F103-1-2-1-1B–Pop2 | |||||||||
| qCR2-137 | 2 | 414 | 2.54 | 2.38 | 0.00 | 0.18 | 39.5 | S2_158674609 | S2_136562142 |
| qCR3-8 | 3 | 262 | 3.33 | 2.97 | 0.63 | 0.52 | S3_8300745 | S3_8888914 | |
| qCR3-151 | 3 | 405 | 6.56 | 6.28 | 0.17 | −0.14 | S3_150831482 | S3_166811360 | |
| qCR4-198 | 4 | 351 | 2.58 | 5.64 | −0.19 | −0.05 | S4_197820294 | S4_200964285 | |
| qCR4-198 | 4 | 354 | 5.77 | 8.85 | 0.24 | #x2212;0.01 | S4_200964285 | S4_198430250 | |
| qCR5-51 | 5 | 374 | 2.93 | 3.7 | −0.17 | 0.27 | S5_186678634 | S5_51355494 | |
| qCR8-123 | 8 | 165 | 4.69 | 6.23 | 0.20 | 0.00 | S8_130213071 | S8_123469991 | |
| qCR9-118 | 9 | 291 | 2.55 | 8.46 | −0.23 | 0.03 | S9_120748383 | S9_118065757 | |
| qCR9-12 | 9 | 359 | 5.63 | 10.95 | −0.28 | −0.03 | S9_12599819 | S9_11929364 | |
| CZL00009 × CZL99017–Pop3 | |||||||||
| qCR1-18 | 1 | 394 | 3 | 5.21 | −0.13 | −0.08 | 12.99 | S1_19328973 | S1_17679542 |
| qCR1-172 | 1 | 501 | 2.81 | 6.09 | 0.67 | −0.6 | S1_196052894 | S1_171534815 | |
| qCR2-16 | 2 | 93 | 3.61 | 5.84 | 0.7 | −0.47 | S2_16401968 | S2_181538947 | |
| qCR9-117 | 9 | 300 | 4.31 | 8.35 | −0.18 | −0.04 | S9_122035011 | S9_116948078 | |
| CML505 × CZL99017–Pop4 | |||||||||
| qCR1-139 | 1 | 182 | 3.87 | 20.16 | 0.32 | 0.67 | 23.89 | S1_139463362 | S1_227241027 |
| qCR4-171 | 4 | 239 | 4.83 | 5.55 | −0.23 | 0.01 | S4_171215058 | S4_173802342 | |
| qCR7-137 | 7 | 87 | 9.79 | 11.01 | −0.34 | 0.22 | S7_140894965 | S7_137169719 | |
| qCR9-47 | 9 | 410 | 3.03 | 2.79 | 0.11 | −0.24 | S9_47064183 | S9_58143264 | |
| qCR9-90 | 9 | 432 | 3.18 | 3 | −0.13 | −0.2 | S9_90366846 | S9_97737243 | |
| CZL0723 × CZL0724–Pop5 | |||||||||
| qCR1-77 | 1 | 89 | 6.52 | 17.39 | 0.29 | −0.09 | 14.28 | S1_73375502 | S1_77145631 |
| Marker | QTL_Name a | Chrom | Pos | α-effect | p Value | PVE (%) | PG |
|---|---|---|---|---|---|---|---|
| S1_77801418 | qCR1_78 | 1 | 77.80 | 0.16 | 1.34 × 10−12 | 2.9 | 7.2 |
| S1_227241027 | qCR1_227 | 1 | 227.24 | 0.17 | 3.75 × 10−7 | 1.5 | 3.8 |
| S2_20589802 | qCR2_20 | 2 | 205.90 | 0.11 | 1.51 × 10−3 | 0.6 | 1.5 |
| S3_172332492 | qCR3_172 | 3 | 172.33 | −0.06 | 1.50 × 10−2 | 0.3 | 0.7 |
| S3_186725598 | qCR3_186 | 3 | 186.73 | 0.10 | 9.88 × 10−3 | 0.4 | 1 |
| S4_828312 | qCR4_1 | 4 | 0.83 | −0.19 | 6.02 × 10−10 | 2.2 | 5.5 |
| S4_5238963 | qCR4_5 | 4 | 5.24 | 0.12 | 7.45 × 10−8 | 1.6 | 4 |
| S4_171215058 | qCR4_171 | 4 | 171.22 | −0.05 | 3.79 × 10−2 | 0.2 | 0.5 |
| S5_2363546 | qCR5_2 | 5 | 2.36 | −0.06 | 2.03 × 10−2 | 0.3 | 0.7 |
| S6_32969273 | qCR6_32 | 6 | 32.97 | 0.10 | 4.49 × 10−5 | 0.9 | 2.2 |
| S6_144280146 | qCR6_144 | 6 | 144.28 | 0.06 | 2.57 × 10−2 | 0.3 | 0.7 |
| S6_154981658 | qCR6_155 | 6 | 154.98 | −0.19 | 8.37 × 10−4 | 0.6 | 1.5 |
| S7_10651847 | qCR7_10 | 7 | 10.65 | −0.28 | 1.78 × 10−13 | 3.1 | 7.8 |
| S7_13389227 | qCR7_13 | 7 | 133.89 | 0.20 | 3.25 × 10−11 | 2.5 | 6.2 |
| S7_137335046 | qCR7_137 | 7 | 137.34 | −0.14 | 3.26 × 10−9 | 2 | 5 |
| S9_134919722 | qCR9_135 | 9 | 134.92 | −0.14 | 5.08 × 10−5 | 0.9 | 2.2 |
| S10_132612571 | qCR10_132 | 10 | 132.61 | −0.15 | 2.76 × 10−9 | 2 | 5 |
| S10_133744261 | qCR10_133 | 10 | 133.74 | −0.09 | 1.79 × 10−3 | 0.5 | 1.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibe, M.; Nyaga, C.; Nair, S.K.; Beyene, Y.; Das, B.; M, S.L.; Bright, J.M.; Makumbi, D.; Kinyua, J.; Olsen, M.S.; et al. Combination of Linkage Mapping, GWAS, and GP to Dissect the Genetic Basis of Common Rust Resistance in Tropical Maize Germplasm. Int. J. Mol. Sci. 2020, 21, 6518. https://doi.org/10.3390/ijms21186518
Kibe M, Nyaga C, Nair SK, Beyene Y, Das B, M SL, Bright JM, Makumbi D, Kinyua J, Olsen MS, et al. Combination of Linkage Mapping, GWAS, and GP to Dissect the Genetic Basis of Common Rust Resistance in Tropical Maize Germplasm. International Journal of Molecular Sciences. 2020; 21(18):6518. https://doi.org/10.3390/ijms21186518
Chicago/Turabian StyleKibe, Maguta, Christine Nyaga, Sudha K. Nair, Yoseph Beyene, Biswanath Das, Suresh L. M, Jumbo M. Bright, Dan Makumbi, Johnson Kinyua, Michael S. Olsen, and et al. 2020. "Combination of Linkage Mapping, GWAS, and GP to Dissect the Genetic Basis of Common Rust Resistance in Tropical Maize Germplasm" International Journal of Molecular Sciences 21, no. 18: 6518. https://doi.org/10.3390/ijms21186518
APA StyleKibe, M., Nyaga, C., Nair, S. K., Beyene, Y., Das, B., M, S. L., Bright, J. M., Makumbi, D., Kinyua, J., Olsen, M. S., Prasanna, B. M., & Gowda, M. (2020). Combination of Linkage Mapping, GWAS, and GP to Dissect the Genetic Basis of Common Rust Resistance in Tropical Maize Germplasm. International Journal of Molecular Sciences, 21(18), 6518. https://doi.org/10.3390/ijms21186518