Molecular Modeling of Pathogenic Mutations in the Keratin 1B Domain



Abstract
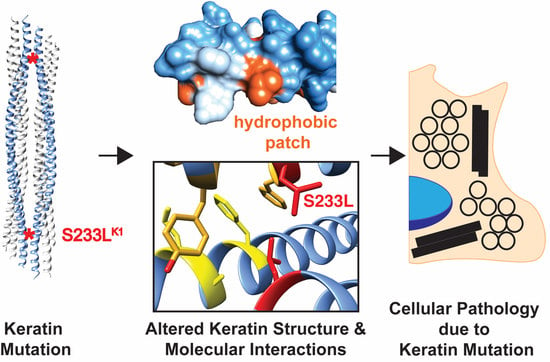
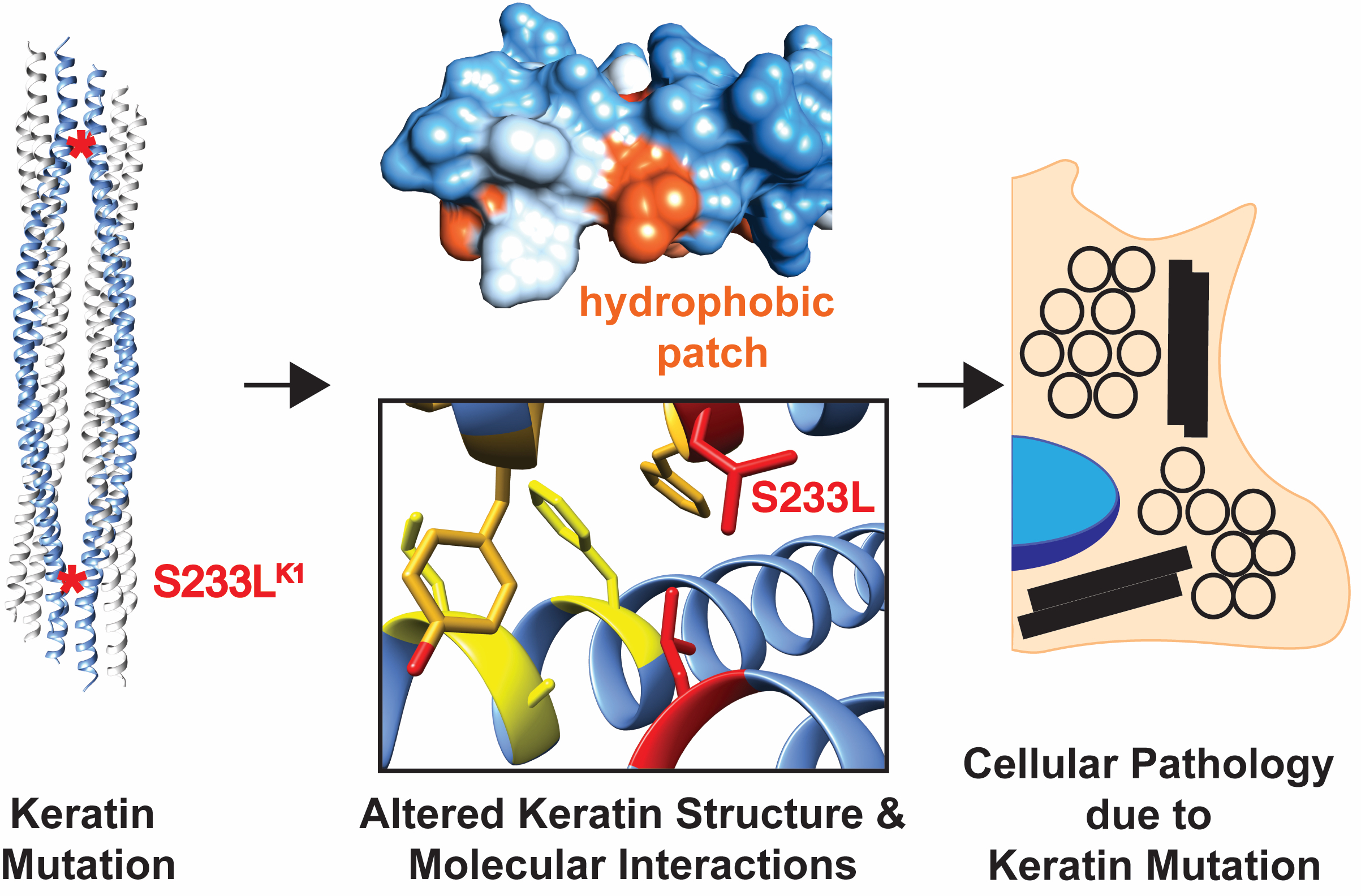
:
1. Introduction
2. Results
2.1. Identification of Keratin 1B Mutations
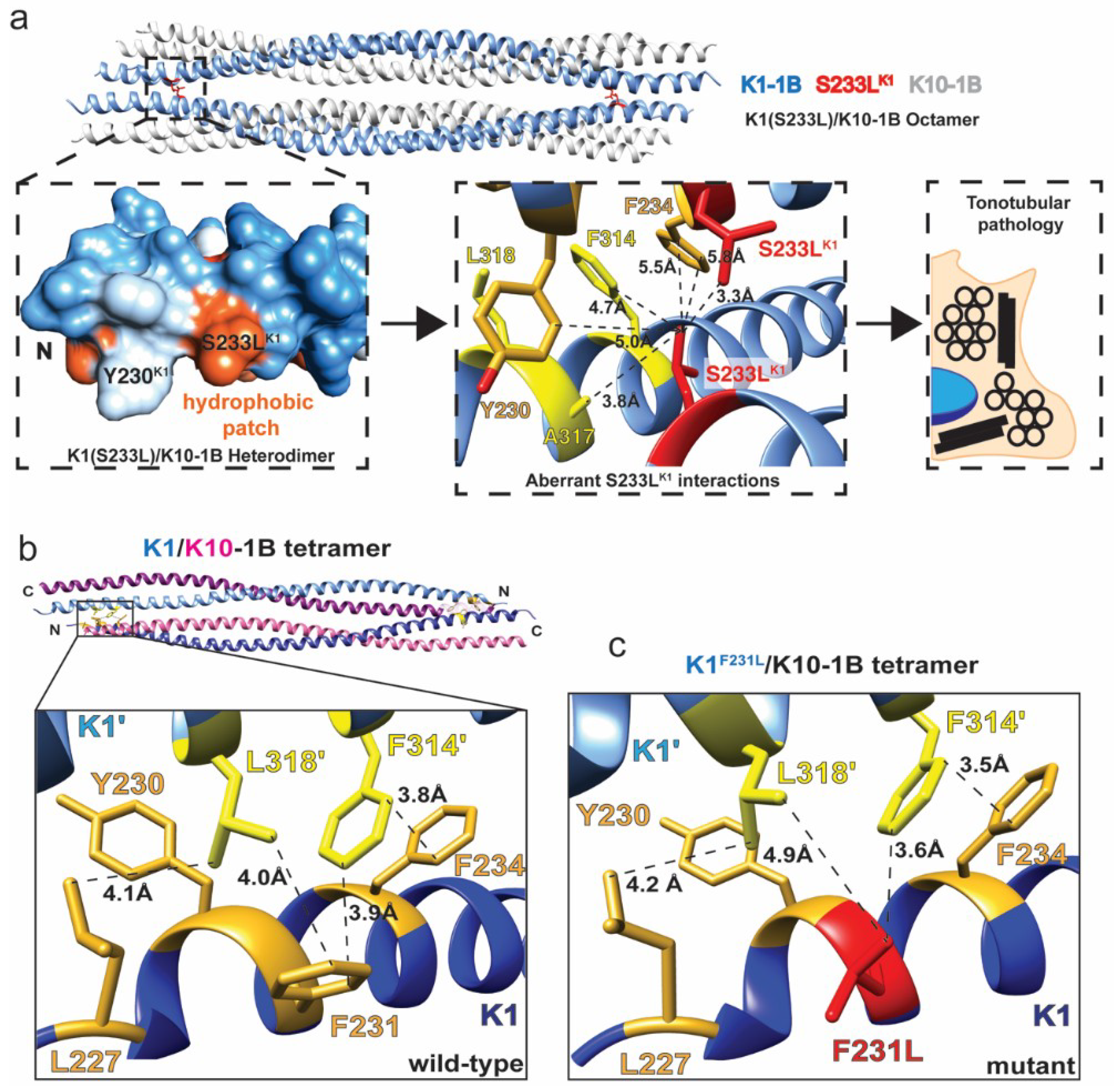
2.2. K1/K10 1B Missense Mutations Associated with Keratodermas
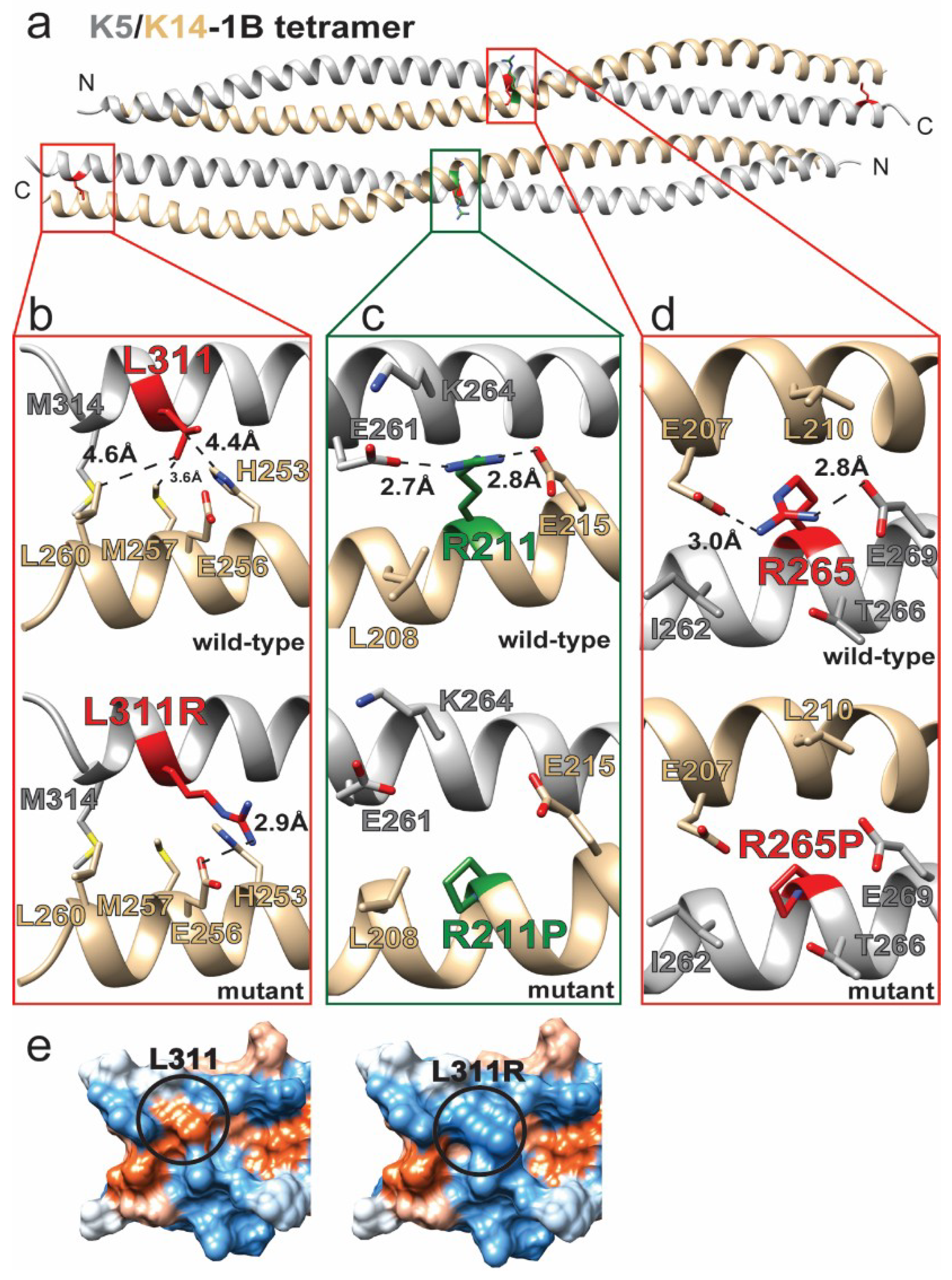
2.3. K5/K14 1B Missense Mutations Cause Epidermolysis Bullosa Simplex
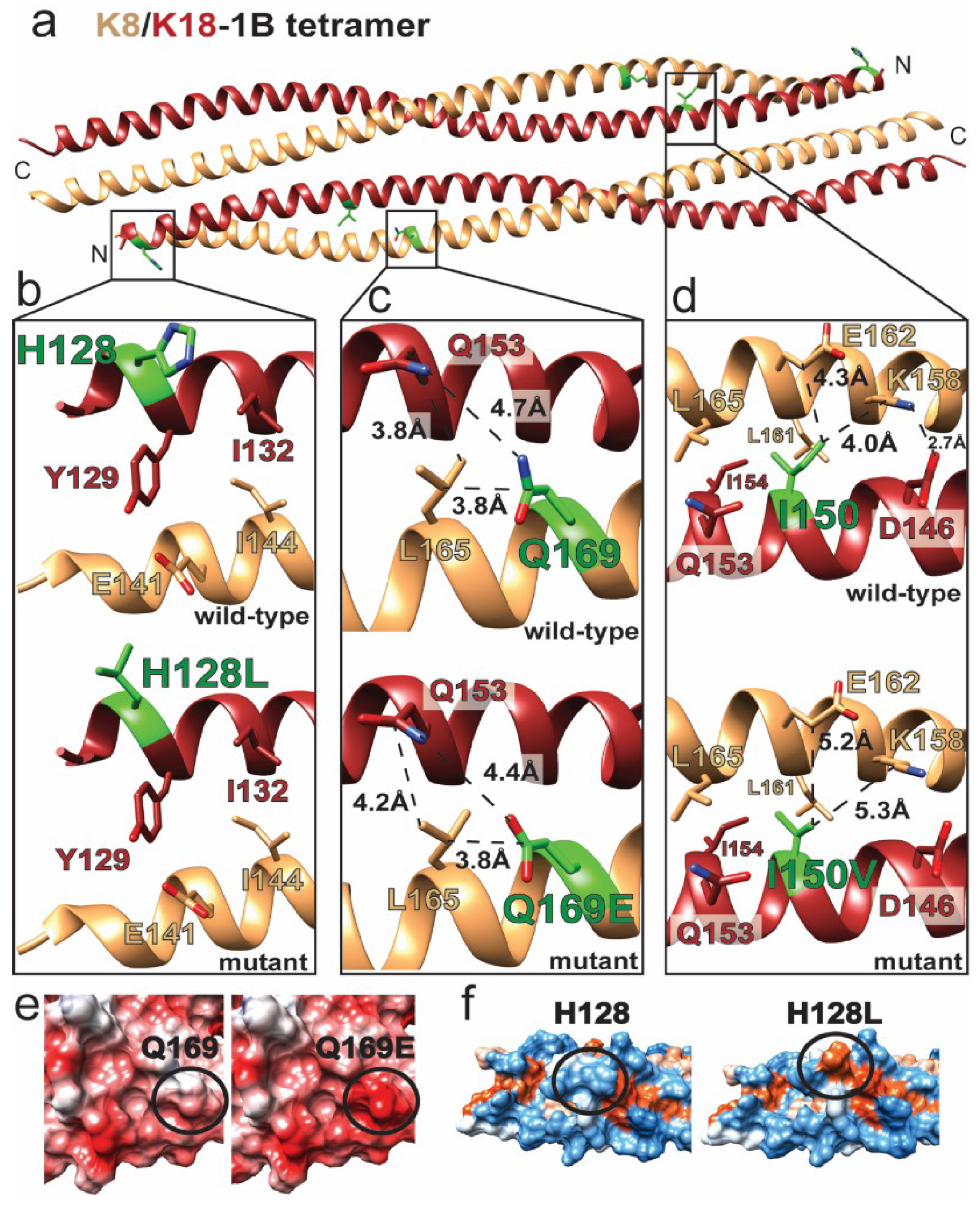
2.4. Structural Alterations in K8/K18 1B Are Associated with Liver Disease
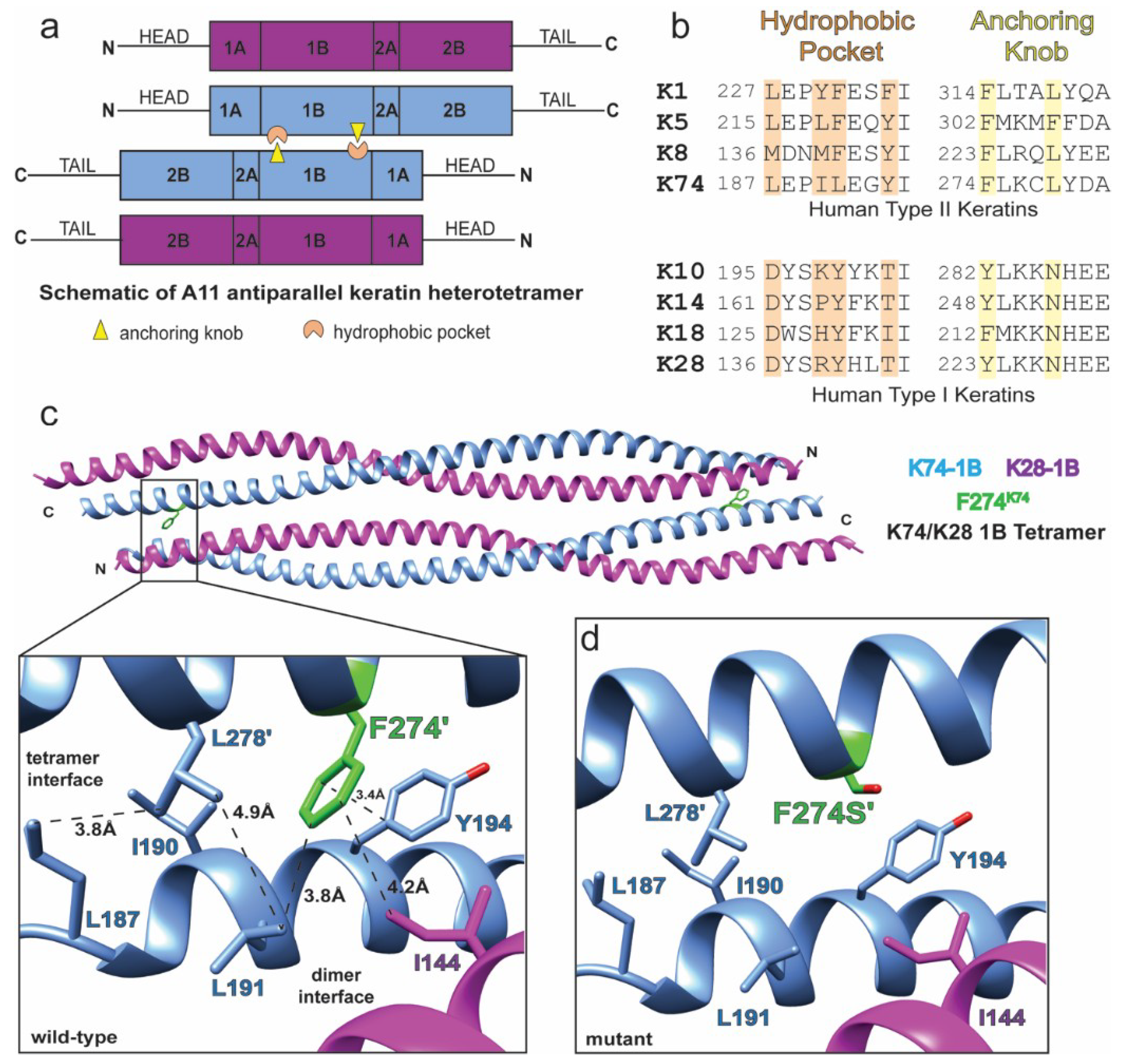
2.5. Anchoring Knob Mutation in the Hard Keratin K74
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IF | Intermediate filament |
| K | Keratin |
| EBS | Epidermolysis bullosa simplex |
| EPPK | Epidermolytic palmoplantar keratoderma |
| PDB | Protein Data Bank |
| HIFD | Human Intermediate Filament Database |
| MALS | Multi-angle light scattering |
| PHNED | Pure hair and nail ectodermal dysplasia |
| NEPPK | Non-epidermolytic palmoplantar keratoderma |
| GFAP | Glial fibrillary acidic protein |
| NCBI | National Center for Biotechnology Information |
References
- Ku, N.O.; Zhou, X.; Toivola, D.M.; Omary, M.B. The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. 1999, 277, 1108–1137. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Bär, H.; Kreplak, L.; Strelkov, S.V.; Aebi, U. Intermediate filaments: From cell architecture to nanomechanics. Nat. Rev. Mol. Cell. Biol. 2007, 8, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, P.A.; Fuchs, E. Elucidating the early stages of keratin filament assembly. J. Cell. Biol. 1990, 111, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.T.; Coulombe, P.A.; Kwan, R.; Omary, M.B. Types I and II Keratin Intermediate Filaments. Cold Spring Harb. Perspect. Biol. 2018, 10, a018275. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.B. “IF-pathies”: A broad spectrum of intermediate filament-associated diseases. J. Clin. Investig. 2009, 119, 1756–1762. [Google Scholar] [CrossRef] [Green Version]
- Szeverenyi, I.; Cassidy, A.J.; Chung, C.W.; Lee, B.T.; Common, J.E.; Ogg, S.C.; Chen, H.; Sim, S.Y.; Goh, W.L.; Ng, K.W.; et al. The Human Intermediate Filament Database: Comprehensive information on a gene family involved in many human diseases. Hum. Mutat. 2008, 29, 351–360. [Google Scholar] [CrossRef]
- Toivola, D.M.; Boor, P.; Alam, C.; Strnad, P. Keratins in health and disease. Curr. Opin. Cell. Biol. 2015, 32, 73–81. [Google Scholar] [CrossRef]
- Steinert, P.M.; Bale, S.J. Genetic skin diseases caused by mutations in keratin intermediate filaments. Trends Genet. 1993, 9, 280–284. [Google Scholar] [CrossRef]
- Jones, L.N.; Steinert, P.M. Hair keratinization in health and disease. Dermatol. Clin. 1996, 14, 633–650. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Siddiqui, I.A.; Syed, D.N.; Adhami, V.M.; Liovic, M.; Mukhtar, H. Keratin gene mutations in disorders of human skin and its appendages. Arch. Biochem. Biophys. 2011, 508, 123–137. [Google Scholar] [CrossRef] [Green Version]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunick, C.G.; Milstone, L.M. The X-ray Crystal Structure of the Keratin 1-Keratin 10 Helix 2B Heterodimer Reveals Molecular Surface Properties and Biochemical Insights into Human Skin Disease. J. Investig. Dermatol. 2017, 137, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Kim, M.S.; Chung, B.M.; Leahy, D.J.; Coulombe, P.A. Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 2012, 19, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomakin, I.B.; Hinbest, A.J.; Ho, M.; Eldirany, S.A.; Bunick, C.G. Crystal structure of keratin 1/10(C401A) 2B heterodimer demonstrates a proclivity for the C-terminus of helix 2B to form higher order molecular contacts. Yale J. Biol. Med. 2020, 93, 3–17. [Google Scholar] [PubMed]
- Eldirany, S.A.; Ho, M.; Bunick, C.G. The Interface between Keratin Structurotype and Human Disease. Structure 2020, 28, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Eldirany, S.A.; Ho, M.; Hinbest, A.J.; Lomakin, I.B.; Bunick, C.G. Human keratin 1/10-1B tetramer structures reveal a knob-pocket mechanism in intermediate filament assembly. EMBO J. 2019, 38, e100741. [Google Scholar] [CrossRef]
- Kim, B.; Kim, S.; Jin, M.S. Crystal structure of the human glial fibrillary acidic protein 1B domain. Biochem. Biophys. Res. Commun. 2018, 503, 2899–2905. [Google Scholar] [CrossRef]
- Aziz, A.; Hess, J.F.; Budamagunta, M.S.; Voss, J.C.; Kuzin, A.P.; Huang, Y.J.; Xiao, R.; Montelione, G.T.; FitzGerald, P.G.; Hunt, J.F. The structure of vimentin linker 1 and rod 1B domains characterized by site-directed spin-labeling electron paramagnetic resonance (SDSL-EPR) and X-ray crystallography. J. Biol. Chem. 2012, 287, 28349–28361. [Google Scholar] [CrossRef] [Green Version]
- Pang, A.H.; Obiero, J.M.; Kulczyk, A.W.; Sviripa, V.M.; Tsodikov, O.V. A crystal structure of coil 1B of vimentin in the filamentous form provides a model of a high-order assembly of a vimentin filament. FEBS J. 2018, 285, 2888–2899. [Google Scholar] [CrossRef] [Green Version]
- Lilina, A.V.; Chernyatina, A.A.; Guzenko, D.; Strelkov, S.V. Lateral A. J. Struct. Biol. 2020, 209, 107404. [Google Scholar] [CrossRef]
- Ahn, J.; Jo, I.; Kang, S.M.; Hong, S.; Kim, S.; Jeong, S.; Kim, Y.H.; Park, B.J.; Ha, N.C. Structural basis for lamin assembly at the molecular level. Nat. Commun. 2019, 10, 3757. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimberg, G.; Hausser, I.; Müller, F.B.; Wodecki, K.; Schaffrath, C.; Krieg, T.; Oji, V.; Traupe, H.; Arin, M.J. Novel and recurrent mutations in the 1B domain of keratin 1 in palmoplantar keratoderma with tonotubules. Br. J. Dermatol. 2009, 160, 446–449. [Google Scholar] [CrossRef]
- Terron-Kwiatkowski, A.; van Steensel, M.A.; van Geel, M.; Lane, E.B.; McLean, W.H.; Steijlen, P.M. Mutation S233L in the 1B domain of keratin 1 causes epidermolytic palmoplantar keratoderma with "tonotubular" keratin. J. Investig. Dermatol. 2006, 126, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Hotz, A.; Oji, V.; Bourrat, E.; Jonca, N.; Mazereeuw-Hautier, J.; Betz, R.C.; Blume-Peytavi, U.; Stieler, K.; Morice-Picard, F.; Schonbuchner, I.; et al. Expanding the Clinical and Genetic Spectrum of KRT1, KRT2 and KRT10 Mutations in Keratinopathic Ichthyosis. Acta Derm. Venereol. 2016, 96, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.W.; Lee, J.S.; Kim, S.E.; Oh, S.W.; Kim, S.C. Novel and recurrent mutations in Keratin 5 and 14 in Korean patients with Epidermolysis bullosa simplex. J. Dermatol. Sci. 2010, 57, 90–94. [Google Scholar] [CrossRef]
- Ciubotaru, D.; Bergman, R.; Baty, D.; Indelman, M.; Pfendner, E.; Petronius, D.; Moualem, H.; Kanaan, M.; Ben Amitai, D.; McLean, W.H.; et al. Epidermolysis bullosa simplex in Israel: Clinical and genetic features. Arch. Dermatol. 2003, 139, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Kucukoglu, O.; Lunova, M.; Guldiken, N.; Lienau, T.C.; Stickel, F.; Omary, M.B. Non-coding keratin variants associate with liver fibrosis progression in patients with hemochromatosis. PLoS ONE 2012, 7, e32669. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, J.A.; Hannoush, Z.C.; Vargas, L.G.; Momany, A.; Garcia, C.C.; Murray, J.C.; Dunnwald, M. A Novel non-sense Mutation in Keratin 10 Causes a Familial Case of Recessive Epidermolytic Ichthyosis. Mol. Genet. Genom. Med. 2013, 1, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, M.F.; Heeres, K.; Pas, H.H.; van Luyn, M.J.; Elema, J.D.; Corden, L.D.; Smith, F.J.; McLean, W.H.; Ramaekers, F.C.; Burton, M.; et al. Effects of keratin 14 ablation on the clinical and cellular phenotype in a kindred with recessive epidermolysis bullosa simplex. J. Investig. Dermatol. 1996, 107, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Schuilenga-Hut, P.H.; Scheffer, H.; Pas, H.H.; Nijenhuis, M.; Buys, C.H.; Jonkman, M.F. Partial revertant mosaicism of keratin 14 in a patient with recessive epidermolysis bullosa simplex. J. Investig. Dermatol. 2002, 118, 626–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hut, P.H.; Vlies, P.v.d.; Jonkman, M.F.; Verlind, E.; Shimizu, H.; Buys, C.H.; Scheffer, H. Exempting homologous pseudogene sequences from polymerase chain reaction amplification allows genomic keratin 14 hotspot mutation analysis. J. Investig. Dermatol. 2000, 114, 616–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.; Anton-Lamprecht, I.; Yu, Q.C.; Jackel, A.; Zabel, B.; Ernst, J.P.; Fuchs, E. A human keratin 14 "knockout": The absence of K14 leads to severe epidermolysis bullosa simplex and a function for an intermediate filament protein. Genes Dev. 1994, 8, 2574–2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yiasemides, E.; Trisnowati, N.; Su, J.; Dang, N.; Klingberg, S.; Marr, P.; Melbourne, W.; Tran, K.; Chow, C.W.; Orchard, D.; et al. Clinical heterogeneity in recessive epidermolysis bullosa due to mutations in the keratin 14 gene, KRT14. Clin. Exp. Dermatol. 2008, 33, 689–697. [Google Scholar] [CrossRef]
- Müller, F.B.; Küster, W.; Wodecki, K.; Almeida, H.; Bruckner-Tuderman, L.; Krieg, T.; Korge, B.P.; Arin, M.J. Novel and recurrent mutations in keratin KRT5 and KRT14 genes in epidermolysis bullosa simplex: Implications for disease phenotype and keratin filament assembly. Hum. Mutat. 2006, 27, 719–720. [Google Scholar] [CrossRef]
- Bolling, M.C.; Lemmink, H.H.; Jansen, G.H.L.; Jonkman, M.F. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br. J. Dermatol. 2011, 164, 637–644. [Google Scholar] [CrossRef]
- Lanschuetzer, C.M.; Klausegger, A.; Pohla-Gubo, G.; Hametner, R.; Richard, G.; Uitto, J.; Hintner, H.; Bauer, J.W. A novel homozygous nonsense deletion/insertion mutation in the keratin 14 gene (Y248X; 744delC/insAG) causes recessive epidermolysis bullosa simplex type Köbner. Clin. Exp. Dermatol. 2003, 28, 77–79. [Google Scholar] [CrossRef]
- García, M.; Santiago, J.L.; Terron, A.; Hernandez-Martin, A.; Vicente, A.; Fortuny, C.; De Lucas, R.; Lopez, J.C.; Cuadrado-Corrales, N.; Holguin, A.; et al. Two novel recessive mutations in KRT14 identified in a cohort of 21 Spanish families with epidermolysis bullosa simplex. Br. J. Dermatol. 2011, 165, 683–692. [Google Scholar] [CrossRef]
- Ku, N.O.; Wright, T.L.; Terrault, N.A.; Gish, R.; Omary, M.B. Mutation of human keratin 18 in association with cryptogenic cirrhosis. J. Clin. Investig. 1997, 99, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Ku, N.O.; Lim, J.K.; Krams, S.M.; Esquivel, C.O.; Keeffe, E.B.; Wright, T.L.; Parry, D.A.; Omary, M.B. Keratins as susceptibility genes for end-stage liver disease. Gastroenterology 2005, 129, 885–893. [Google Scholar] [CrossRef]
- Raykova, D.; Klar, J.; Azhar, A.; Khan, T.N.; Malik, N.A.; Iqbal, M.; Tariq, M.; Baig, S.M.; Dahl, N. Autosomal recessive transmission of a rare KRT74 variant causes hair and nail ectodermal dysplasia: Allelism with dominant woolly hair/hypotrichosis. PLoS ONE 2014, 9, e93607. [Google Scholar] [CrossRef]
- Wevers, A.; Kuhn, A.; Mahrle, G. Palmoplantar keratoderma with tonotubular keratin. J. Am. Acad. Dermatol. 1991, 24, 638–642. [Google Scholar] [CrossRef]
- Woolfson, D.N.; Williams, D.H. The influence of proline residues on alpha-helical structure. FEBS Lett. 1990, 277, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Strnad, P.; Paschke, S.; Jang, K.H.; Ku, N.O. Keratins: Markers and modulators of liver disease. Curr. Opin. Gastroenterol. 2012, 28, 209–216. [Google Scholar] [CrossRef]
- Ku, N.O.; Darling, J.M.; Krams, S.M.; Esquivel, C.O.; Keeffe, E.B.; Sibley, R.K.; Lee, Y.M.; Wright, T.L.; Omary, M.B. Keratin 8 and 18 mutations are risk factors for developing liver disease of multiple etiologies. Proc. Natl. Acad. Sci. USA. 2003, 100, 6063–6068. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, Y.; Wajid, M.; Petukhova, L.; Kurban, M.; Christiano, A.M. Autosomal-dominant woolly hair resulting from disruption of keratin 74 (KRT74), a potential determinant of human hair texture. Am. J. Hum. Genet. 2010, 86, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Langbein, L.; Rogers, M.A.; Praetzel-Wunder, S.; Helmke, B.; Schirmacher, P.; Schweizer, J. K25 (K25irs1), K26 (K25irs2), K27 (K25irs3), and K28 (K25irs4) represent the type I inner root sheath keratins of the human hair follicle. J. Investig. Dermatol. 2006, 126, 2377–2386. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Kim, M.S.; Li, S.; Leahy, D.J.; Coulombe, P.A. Structure-Function Analyses of a Keratin Heterotypic Complex Identify Specific Keratin Regions Involved in Intermediate Filament Assembly. Structure 2020, 28, 355–362. [Google Scholar] [CrossRef]
- Bernot, K.M.; Lee, C.H.; Coulombe, P.A. A small surface hydrophobic stripe in the coiled-coil domain of type I keratins mediates tetramer stability. J. Cell. Biol. 2005, 168, 965–974. [Google Scholar] [CrossRef]
- Sun, J.; Groppi, V.E.; Gui, H.; Chen, L.; Xie, Q.; Liu, L.; Omary, M.B. High-Throughput Screening for Drugs that Modulate Intermediate Filament Proteins. Methods Enzymol. 2016, 568, 163–185. [Google Scholar] [CrossRef] [Green Version]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar]
- Landrum, M.J.; Kattman, B.L. ClinVar at five years: Delivering on the promise. Hum. Mutat. 2018, 39, 1623–1630. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Mutation | Protein Mutation | Protein | Hetero Pair | Mutation Type | PolyphnScore | PolyphenPrediction | Disease Association | Disease Mechanism | References |
|---|---|---|---|---|---|---|---|---|---|
| c.693T>G | p.Phe231Leu | K1 | K1/10 | Missense | 0.971 | Probably Damaging | NEPPK | Disrupts 1B knob-pocket interaction | [23] |
| c.698C>T | p.Ser233Leu | K1 | K1/10 | Missense | 0.704 | Possibly Damaging | EPPK, NEPPK, BCIE/EHK | Tonotubular filament formation | [16,23,24,25] |
| c.794G>C | p.Arg265Pro | K5 | K5/14 | Missense | 0.975 | Probably Damaging | EBS-K | Unknown | [26] |
| c.932T>G | p.Leu311Arg | K5 | K5/14 | Missense | 0.908 | Possibly Damaging | EBS-WC | Unknown | [27] |
| c.475C>G | p.Gln169Glu | K8 | K8/18 | Missense | 0.171 | Benign | Cryptogenic cirrhosis | Unknown | [28] |
| c.846T>A | p.Tyr282X | K10 | K1/10 | Nonsense (truncation) | N/A | N/A | CIEH | LOF (K10 K/O), aggregated K1, compensatory upregulation of K14 & K17 | [29] |
| c.526-2A>C | p.[Ile176ValfsX2, Ile176ProfsX30] | K14 | K5/14 | Frame-shift (truncation) | N/A | N/A | REBS, REBS-K | Basal K14 IF loss, compensatory K15 protofilaments | [30,31,32] |
| c.[612T>A]+[612T>A] | p.[Tyr204X]+[Tyr204X] | K14 | K5/14 | Nonsense (truncation) | N/A | N/A | REBS-K | "Natural K14 K/O", Basal K14 IF loss, insoluble keratin aggregation | [33,34] |
| c.632G>C | p.Arg211Pro | K14 | K5/14 | Missense | 0.999 | Probably Damaging | EBS-WC | Unknown | [35] |
| c.740_748delCCTACCTGAinsGAA | p.Ala247_Lys250delinsGlu | K14 | K5/14 | Indel (in-frame) | N/A | N/A | EBS-WC | Unknown | [36] |
| c.744delCinsAG | p.Tyr248X | K14 | K5/14 | Nonsense (truncation) | N/A | N/A | REBS-K | "Natural K14 K/O", Basal K14 IF loss, insoluble keratin aggregation | [37] |
| c.749delA | p.Lys250ArgfsX8 | K14 | K5/14 | Deletion (frame-shift) | N/A | N/A | REBS | Absent K14 expression | [38] |
| c.383A>T | p.His128Leu | K18 | K8/18 | Missense | 0.014 | Benign | Cryptogenic cirrhosis | Abnormal IF assembly | [39] |
| c.448A>G | p.Ile150Val | K18 | K8/18 | Missense | 0.247 | Benign | Liver disease | Unknown | [40] |
| c.821T>C | p.Phe274Ser | K74 | K74/14 or K74/28 | Missense | 0.998 | Probably Damaging | Ectodermal Dysplasia, Pure Hair-Nail Type, ADWH | Disrupts 1B knob-pocket interaction | [16,41] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinbest, A.J.; Eldirany, S.A.; Ho, M.; Bunick, C.G. Molecular Modeling of Pathogenic Mutations in the Keratin 1B Domain. Int. J. Mol. Sci. 2020, 21, 6641. https://doi.org/10.3390/ijms21186641
Hinbest AJ, Eldirany SA, Ho M, Bunick CG. Molecular Modeling of Pathogenic Mutations in the Keratin 1B Domain. International Journal of Molecular Sciences. 2020; 21(18):6641. https://doi.org/10.3390/ijms21186641
Chicago/Turabian StyleHinbest, Alexander J., Sherif A. Eldirany, Minh Ho, and Christopher G. Bunick. 2020. "Molecular Modeling of Pathogenic Mutations in the Keratin 1B Domain" International Journal of Molecular Sciences 21, no. 18: 6641. https://doi.org/10.3390/ijms21186641
APA StyleHinbest, A. J., Eldirany, S. A., Ho, M., & Bunick, C. G. (2020). Molecular Modeling of Pathogenic Mutations in the Keratin 1B Domain. International Journal of Molecular Sciences, 21(18), 6641. https://doi.org/10.3390/ijms21186641