Atypical Chronic Myeloid Leukemia: Where Are We Now?

 ,
,  and
and
Abstract
:1. General Concepts
2. Clinical Case 1: aCML in Younger Patients
3. Clinical Case 2: aCML in the Elderly Patient/Unfit for Allogeneic Stem Cell Transplantation
4. Characteristics of aCML at Presentation and Prognosis
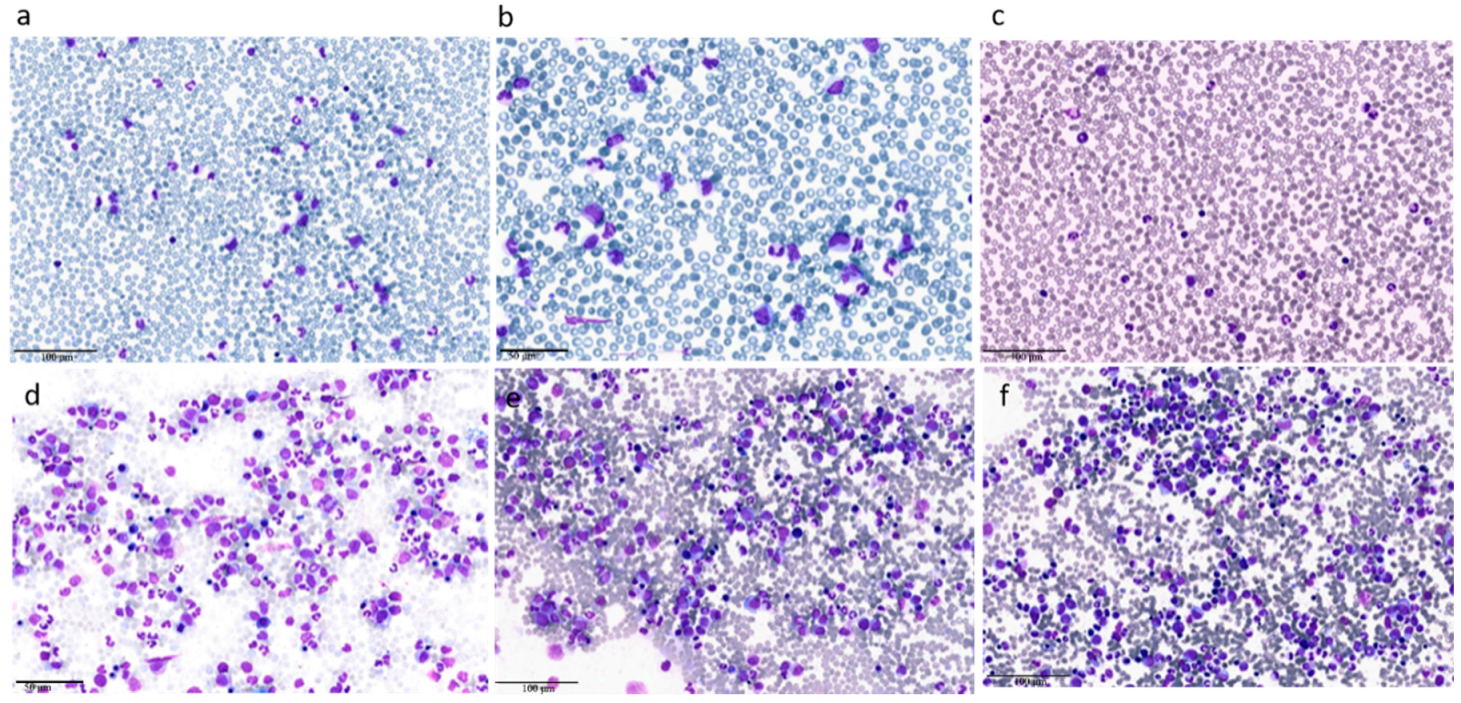
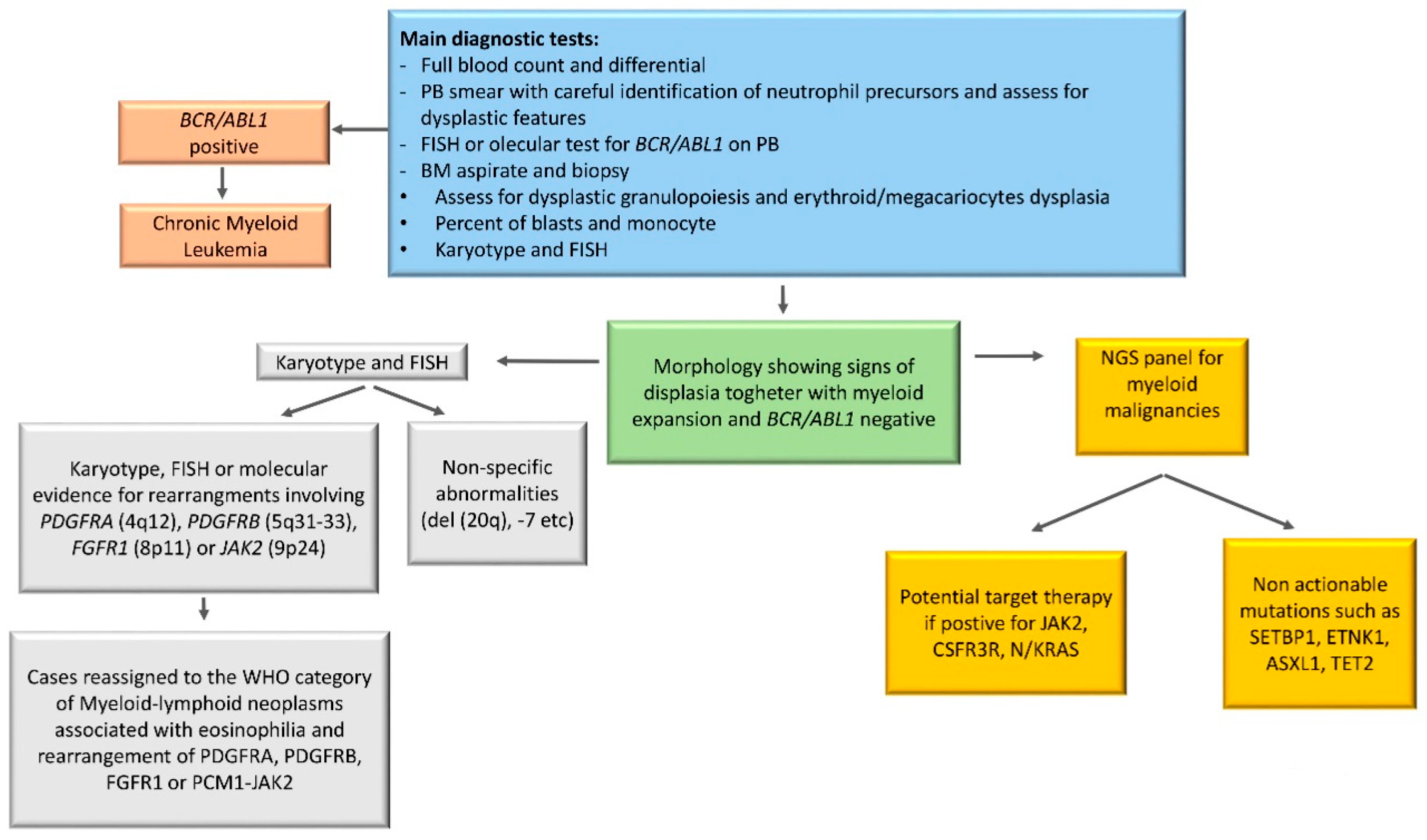
5. Diagnostic Tools in aCML
6. Cytogenetics in aCML
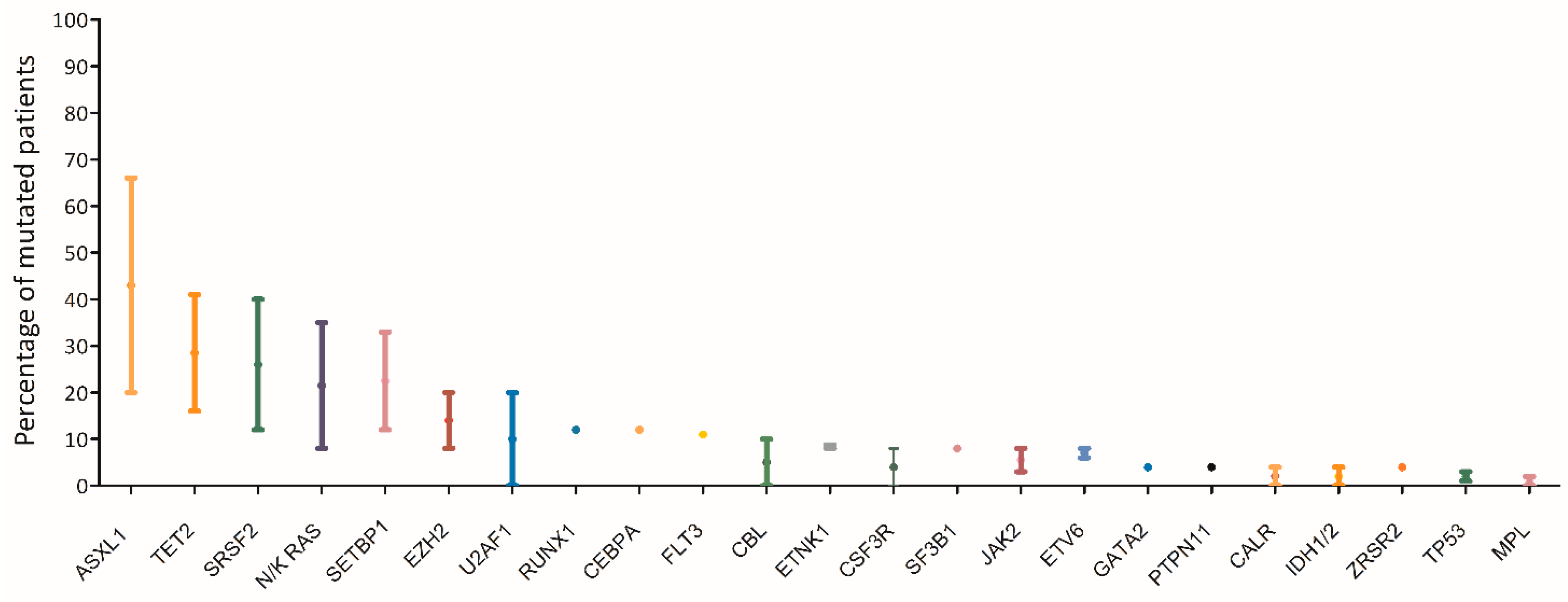
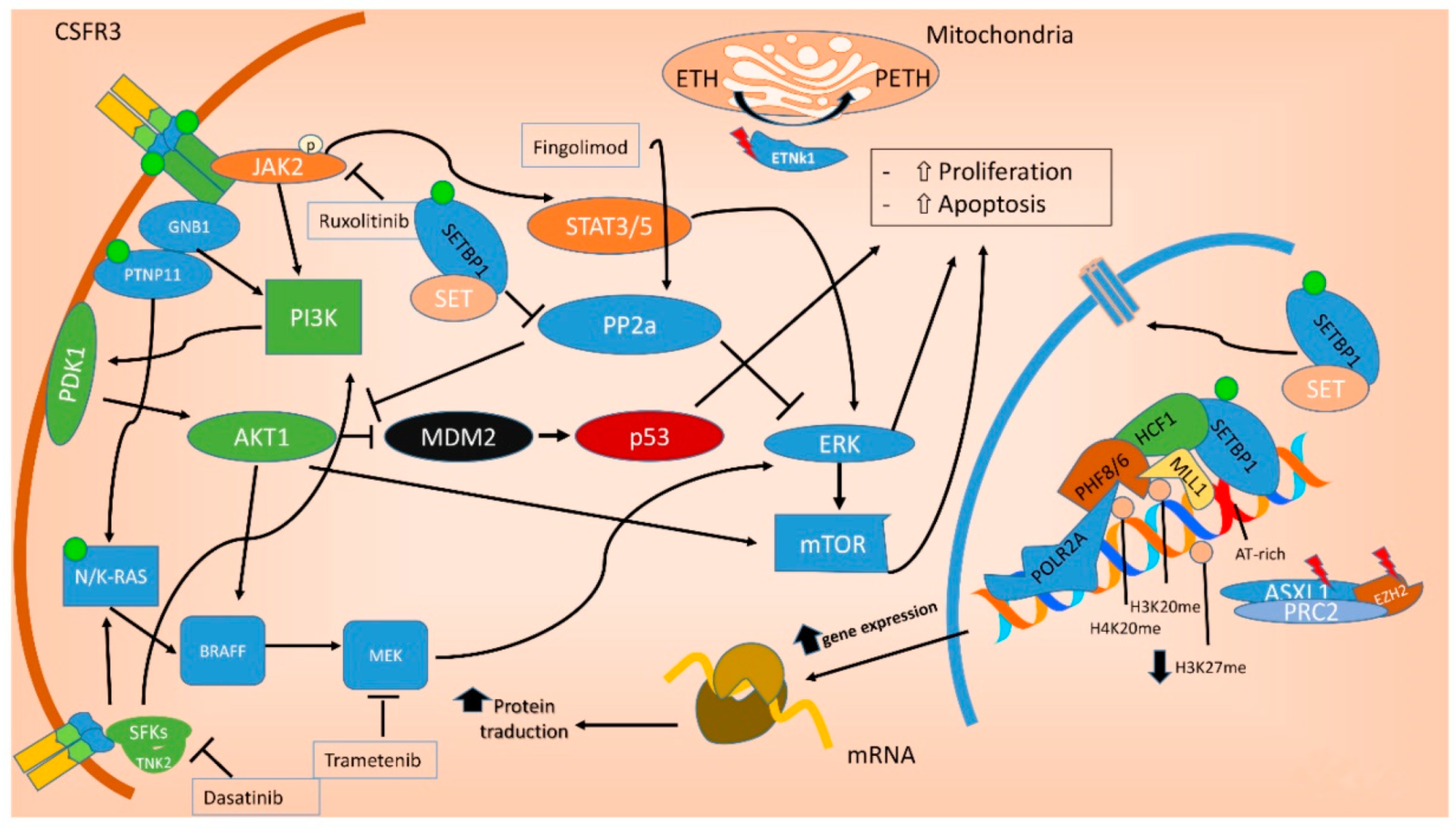
7. Molecular Landscape of aCML
8. Clinical Case 1: Treatment Choices in a Transplant Eligible Patient
9. Clinical Case 2: Treatment Approach in a Transplant Ineligible Patient
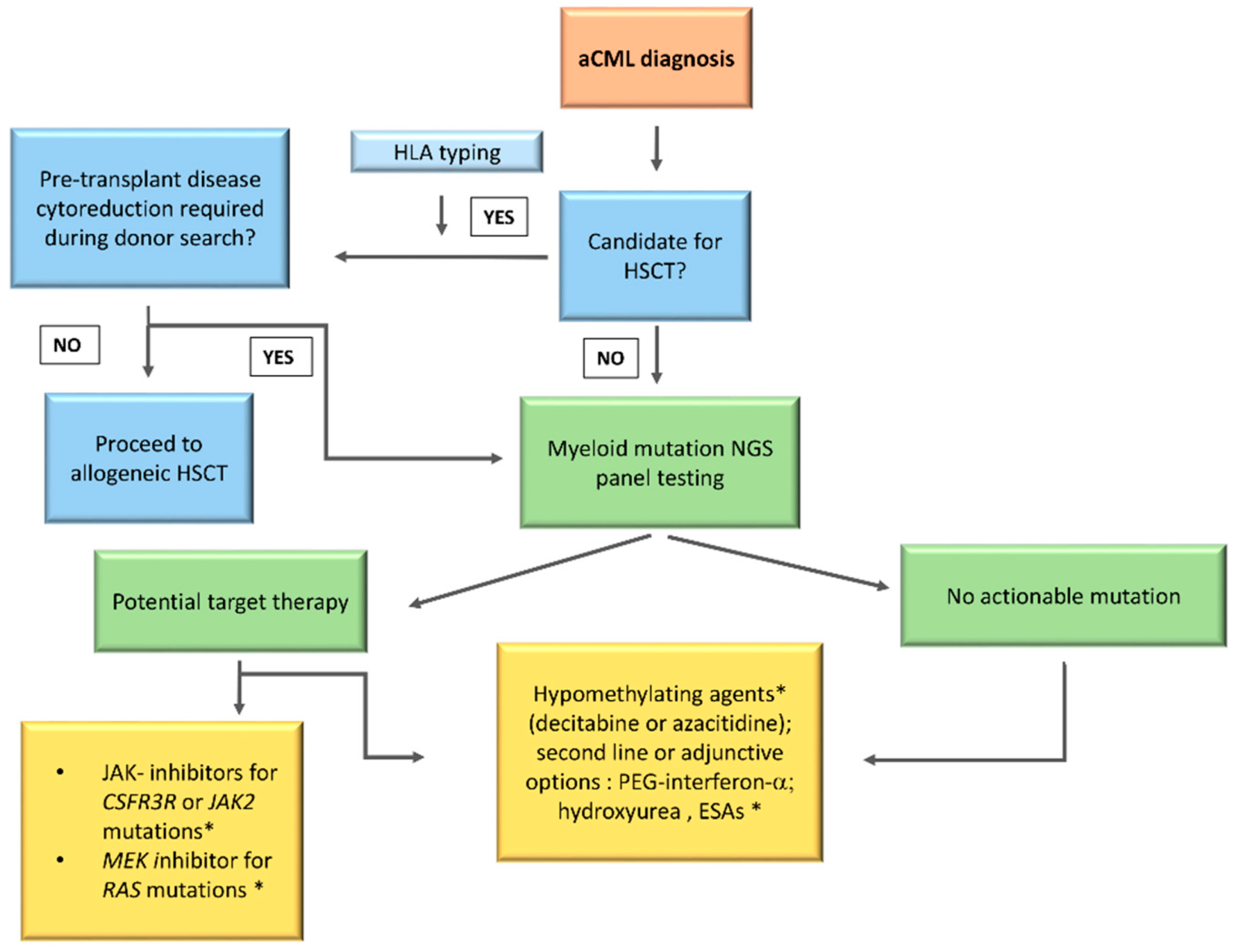
10. Treatment
10.1. Hematopoietic Stem Cell Transplant
10.2. Hypomethylating Agents
10.3. AML-Like Chemotherapy
10.4. Interferon-Alpha and Hydroxyurea
10.5. Target Therapy
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gambacorti-Passerini, C.B.; Donadoni, C.; Parmiani, A.; Pirola, A.; Redaelli, S.; Signore, G.; Piazza, V.; Malcovati, L.; Fontana, D.; Spinelli, R.; et al. Recurrent ETNK1 mutations in atypical chronic myeloid leukemia. Blood 2015, 125, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, M.M.; Barraco, D.; Lasho, T.L.; Finke, C.M.; Reichard, K.; Hoversten, K.P.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Targeted next generation sequencing and identification of risk factors in World Health Organization defined atypical chronic myeloid leukemia. Am. J. Hematol. 2017, 92, 542–548. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Kurzrock, R.; Bueso-Ramos, C.E.; Kantarjian, H.; Freireich, E.; Tucker, S.L.; Siciliano, M.; Pilat, S.; Talpaz, M. BCR Rearrangement–Negative Chronic Myelogenous Leukemia Revisited. J. Clin. Oncol. 2001, 19, 2915–2926. [Google Scholar] [CrossRef]
- Dao, K.-H.T.; Tyner, J.W. What’s different about atypical CML and chronic neutrophilic leukemia? Hematol. Am. Soc. Hematol. Educ. Progr. 2015, 2015, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Thakral, B.; Anastasi, J.; Wang, S.A. Myeloproliferative and “Overlap” Myelodysplastic/Myeloproliferative Neoplasms. Hematopathology 2018, 488–538. [Google Scholar] [CrossRef]
- Wang, S.A.; Hasserjian, R.P.; Fox, P.S.; Rogers, H.J.; Geyer, J.T.; Chabot-Richards, D.; Weinzierl, E.; Hatem, J.; Jaso, J.; Kanagal-Shamanna, R.; et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood 2014, 123, 2645–2651. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Maxson, J.E.; George, T.I.; Tyner, J.W. The new genetics of chronic neutrophilic leukemia and atypical CML: Implications for diagnosis and treatment. Blood 2013, 122, 1707–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breccia, M.; Biondo, F.; Latagliata, R.; Carmosino, I.; Mandelli, F.; Alimena, G. Identification of risk factors in atypical chronic myeloid leukemia. Haematologica 2006, 91, 1566–1568. [Google Scholar]
- Martiat, P.; Michaux, J.L.; Rodhain, J. Philadelphia-negative (Ph−) chronic myeloid leukemia (CML): Comparison with Ph+ CML and chronic myelomonocytic leukemia. The Groupe Français de Cytogénétique Hématologique. Blood 1991, 78, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Drozd-Sokołowska, J.; Mądry, K.; Waszczuk-Gajda, A.; Biecek, P.; Szwedyk, P.; Budziszewska, K.; Raźny, M.; Dutka, M.; Obara, A.; Wasilewska, E.; et al. Atypical chronic myeloid leukaemia: A case of an orphan disease-A multicenter report by the Polish Adult Leukemia Group. Hematol. Oncol. 2018, 36, 570–575. [Google Scholar] [CrossRef]
- Giri, S.; Pathak, R.; Martin, M.G.; Bhatt, V.R. Characteristics and survival of BCR/ABL negative chronic myeloid leukemia: A retrospective analysis of the Surveillance, Epidemiology and End Results database. Ther. Adv. Hematol. 2015, 6, 308–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, P.; Gundabolu, K.; Amador, C.; Rayamajhi, S.; Bhatt, V.R. Atypical chronic myeloid leukemia: A rare entity with management challenges. Futur. Oncol. 2018, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Dewald, G.W.; Lasho, T.L.; Powell, H.L.; McClure, R.F.; Levine, R.L.; Gilliland, D.G.; Tefferi, A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood 2005, 106, 1207–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wilmot, B.; Bottomly, D.; Dao, K.-H.T.; Stevens, E.; Eide, C.A.; Khanna, V.; Rofelty, A.; Savage, S.; Reister Schultz, A.; et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood 2019, 134, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Sadigh, S.; Hasserjian, R.P.; Hobbs, G. Distinguishing atypical chronic myeloid leukemia from other Philadelphia-negative chronic myeloproliferative neoplasms. Curr. Opin. Hematol. 2020, 27, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Meggendorfer, M.; Bacher, U.; Alpermann, T.; Haferlach, C.; Kern, W.; Gambacorti-Passerini, C.; Haferlach, T.; Schnittger, S. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i(17)(q10), ASXL1 and CBL mutations. Leukemia 2013, 27, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J. How I treat atypical chronic myeloid leukemia. Blood 2017, 129, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Lasho, T. Atypical CML- the role of morphology and precision genomics. Best Pract. Res. Clin. Haematol. 2020, 33, 101133. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Haferlach, T.; Alpermann, T.; Jeromin, S.; Haferlach, C.; Kern, W.; Schnittger, S. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica 2014, 99, e244–e246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2013, 45, 18. [Google Scholar] [CrossRef]
- Onida, F.; Ball, G.; Kantarjian, H.M.; Smith, T.L.; Glassman, A.; Albitar, M.; Scappini, B.; Rios, M.B.; Keating, M.J.; Beran, M. Characteristics and outcome of patients with Philadelphia chromosome negative, bcr/abl negative chronic myelogenous leukemia. Cancer 2002, 95, 1673–1684. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.V.; Kreil, S.; Zoi, K.; Waghorn, K.; Curtis, C.; Zhang, L.; Score, J.; Seear, R.; Chase, A.J.; Grand, F.H.; et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005, 106, 2162–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damm, F.; Itzykson, R.; Kosmider, O.; Droin, N.; Renneville, A.; Chesnais, V.; Gelsi-Boyer, V.; de Botton, S.; Vey, N.; Preudhomme, C.; et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia 2013, 27, 1401–1403. [Google Scholar] [CrossRef]
- Zoi, K.; Cross, N.C.P. Molecular pathogenesis of atypical CML, CMML and MDS/MPN-unclassifiable. Int. J. Hematol. 2015, 101, 229–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasho, T.L.; Finke, C.M.; Zblewski, D.; Patnaik, M.; Ketterling, R.P.; Chen, D.; Hanson, C.A.; Tefferi, A.; Pardanani, A. Novel recurrent mutations in ethanolamine kinase 1 (ETNK1) gene in systemic mastocytosis with eosinophilia and chronic myelomonocytic leukemia. Blood Cancer J. 2015, 5, e275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E.; et al. Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Rocca, S.; Carrà, G.; Poggio, P.; Morotti, A.; Brancaccio, M. Targeting few to help hundreds: JAK, MAPK and ROCK pathways as druggable targets in atypical chronic myeloid leukemia. Mol. Cancer 2018, 17, 40. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Schroeder, T.; Zabelina, T.; Badbaran, A.; Bacher, U.; Kobbe, G.; Ayuk, F.; Wolschke, C.; Schnittger, S.; Kohlmann, A.; et al. Postallogeneic monitoring with molecular markers detected by pretransplant next-generation or Sanger sequencing predicts clinical relapse in patients with myelodysplastic/myeloproliferative neoplasms. Eur. J. Haematol. 2014, 92, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; McCarron, S.L.; Haslam, K.; O’Donovan, M.T.; Conneally, E. The CSF3R T618I mutation as a disease-specific marker of atypical CML post allo-SCT. Bone Marrow Transplant. 2014, 49, 843–844. [Google Scholar] [CrossRef] [PubMed]
- Savona, M.R.; Malcovati, L.; Komrokji, R.; Tiu, R.V.; Mughal, T.I.; Orazi, A.; Kiladjian, J.J.; Padron, E.; Solary, E.; Tibes, R.; et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood 2015, 125, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.N.; Lee, J.H.; Lee, J.H.; Kim, D.Y.; Kim, S.D.; Kang, Y.A.; Lee, Y.S.; Lee, K.H. Allogeneic hematopoietic cell transplantation in adult patients with myelodysplastic/myeloproliferative neoplasms. Blood Res. 2013, 48, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Mittal, P.; Saliba, R.M.; Giralt, S.A.; Shahjahan, M.; Cohen, A.I.; Karandish, S.; Onida, F.; Beran, M.; Champlin, R.E.; de Lima, M. Allogeneic transplantation: A therapeutic option for myelofibrosis, chronic myelomonocytic leukemia and Philadelphia-negative/BCR-ABL-negative chronic myelogenous leukemia. Bone Marrow Transplant. 2004, 33, 1005–1009. [Google Scholar] [CrossRef]
- Koldehoff, M.; Beelen, D.W.; Trenschel, R.; Steckel, N.K.; Peceny, R.; Ditschkowski, M.; Ottinger, H.; Elmaagacli, A.H. Outcome of hematopoietic stem cell transplantation in patients with atypical chronic myeloid leukemia. Bone Marrow Transplant. 2004, 34, 1047–1050. [Google Scholar] [CrossRef] [Green Version]
- Koldehoff, M.; Steckel, N.K.; Hegerfeldt, Y.; Ditschkowski, M.; Beelen, D.W.; Elmaagacli, A.H. Clinical course and molecular features in 21 patients with atypical chronic myeloid leukemia. Int. J. Lab. Hematol. 2012, 34, e3–e5. [Google Scholar] [CrossRef]
- Onida, F.; de Wreede, L.C.; van Biezen, A.; Eikema, D.-J.; Byrne, J.L.; Iori, A.P.; Schots, R.; Jungova, A.; Schetelig, J.; Finke, J.; et al. Allogeneic stem cell transplantation in patients with atypical chronic myeloid leukaemia: A retrospective study from the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Br. J. Haematol. 2017, 177, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Talati, C.; Padron, E. An Exercise in Extrapolation: Clinical Management of Atypical CML, MDS/MPN-Unclassifiable, and MDS/MPN-RS-T. Curr. Hematol. Malig. Rep. 2016, 11, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2016 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2016, 91, 631–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; O’Brien, S.; Cortes, J.; Giles, F.J.; Faderl, S.; Issa, J.-P.; Garcia-Manero, G.; Rios, M.B.; Shan, J.; Andreeff, M.; et al. Results of decitabine (5-aza-2′deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer 2003, 98, 522–528. [Google Scholar] [CrossRef]
- Hausmann, H.; Bhatt, V.R.; Yuan, J.; Maness, L.J.; Ganti, A.K. Activity of single-agent decitabine in atypical chronic myeloid leukemia. J. Oncol. Pharm. Pract. 2016, 22, 790–794. [Google Scholar] [CrossRef]
- Mao, L.; You, L.; Yang, M.; Li, Y.; Ye, X.; Tong, H.Y. The First Case of Decitabine Successfully in Treatment of Atypical Chronic Myeloid Leukemia with CEBPA Double Mutation. Chemother. Open Access 2013, 2, 114. [Google Scholar] [CrossRef]
- Tong, X.; Li, J.; Zhou, Z.; Zheng, D.; Liu, J.; Su, C. Efficacy and side-effects of decitabine in treatment of atypical chronic myeloid leukemia. Leuk. Lymphoma 2015, 56, 1911–1913. [Google Scholar] [CrossRef]
- Jiang, H.; Wu, Z.; Ren, L.; Tao, D.; Tong, H. Decitabine for the treatment of atypical chronic myeloid leukemia: A report of two cases. Oncol. Lett. 2016, 11, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Nishihori, T.; Perkins, J.; Mishra, A.; Komrokji, R.; Kim, J.; Kharfan-Dabaja, M.A.; Perez, L.; Lancet, J.; Fernandez, H.; List, A.; et al. Pretransplantation 5-azacitidine in high-risk myelodysplastic syndrome. Biol. Blood Marrow Transplant. 2014, 20, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Gerds, A.T.; Deeg, H.J. Transplantation for myelodysplastic syndrome in the era of hypomethylating agents. Curr. Opin. Hematol. 2012, 19, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Voso, M.T.; Leone, G.; Piciocchi, A.; Fianchi, L.; Santarone, S.; Candoni, A.; Criscuolo, M.; Masciulli, A.; Cerqui, E.; Molteni, A.; et al. Feasibility of allogeneic stem-cell transplantation after azacitidine bridge in higher-risk myelodysplastic syndromes and low blast count acute myeloid leukemia: Results of the BMT-AZA prospective study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H.; Cortes, J.; Thomas, D.; Garcia-Manero, G.; Ferrajoli, A.; Faderl, S.; Richie, M.A.; Beran, M.; Giles, F.; et al. PEG-IFN-α-2b therapy in BCR-ABL–negative myeloproliferative disorders. Cancer 2007, 110, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Maxson, J.E.; Luty, S.B.; Agarwal, A.; Royer, L.R.; Abel, M.L.; MacManiman, J.D.; Loriaux, M.M.; Druker, B.J.; Tyner, J.W. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood 2013, 122, 3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxson, J.E.; Luty, S.B.; MacManiman, J.D.; Paik, J.C.; Gotlib, J.; Greenberg, P.; Bahamadi, S.; Savage, S.L.; Abel, M.L.; Eide, C.A.; et al. The Colony Stimulating Factor 3 Receptor T640N mutation is oncogenic, sensitive to JAK inhibition, and mimics T618I. Clin. Cancer Res. 2016, 22, 757. [Google Scholar] [CrossRef] [Green Version]
- Dao, K.-H.T.; Solti, M.B.; Maxson, J.E.; Winton, E.F.; Press, R.D.; Druker, B.J.; Tyner, J.W. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk. Res. Reports 2014, 3, 67–69. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.L.; Desai, A.V.; Bailey, L.C.; Aplenc, R.; Burnworth, B.; Zehentner, B.K.; Teachey, D.T.; Wertheim, G. Atypical Chronic Myeloid Leukemia in Two Pediatric Patients. Pediatr. Blood Cancer 2016, 63, 156–159. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.M.; Garcia-Manero, G.; Cortes, J.E.; Pemmaraju, N.; Wang, X.; Nogueras-Gonzalez, G.; Jabbour, E.; Bose, P.; Kadia, T.; et al. A phase II trial of ruxolitinib in combination with azacytidine in myelodysplastic syndrome/myeloproliferative neoplasms. Am. J. Hematol. 2018, 93, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Shanavas, M.; Popat, U.; Michaelis, L.C.; Fauble, V.; McLornan, D.; Klisovic, R.; Mascarenhas, J.; Tamari, R.; Arcasoy, M.O.; Davies, J.; et al. Outcomes of Allogeneic Hematopoietic Cell Transplantation in Patients with Myelofibrosis with Prior Exposure to Janus Kinase 1/2 Inhibitors. Biol. Blood Marrow Transplant. 2016, 22, 432–440. [Google Scholar] [CrossRef] [Green Version]
- Shahnaz Syed Abd Kadir, S.; Christopeit, M.; Wulf, G.; Wagner, E.; Bornhauser, M.; Schroeder, T.; Crysandt, M.; Mayer, K.; Jonas, J.; Stelljes, M.; et al. Impact of ruxolitinib pretreatment on outcomes after allogeneic stem cell transplantation in patients with myelofibrosis. Eur. J. Haematol. 2018, 101, 305–317. [Google Scholar] [CrossRef]
- Burgess, M.R.; Hwang, E.; Firestone, A.J.; Huang, T.; Xu, J.; Zuber, J.; Bohin, N.; Wen, T.; Kogan, S.C.; Haigis, K.M.; et al. Preclinical efficacy of MEK inhibition in NRAS-mutant AML. Blood 2014, 124, 3947–3955. [Google Scholar] [CrossRef]
- Borthakur, G.; Popplewell, L.; Boyiadzis, M.; Foran, J.; Platzbecker, U.; Vey, N.; Walter, R.B.; Olin, R.; Raza, A.; Giagounidis, A.; et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer 2016, 122, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, V.; Pierce, S.T.; Dao, K.-H.T.; Tognon, C.E.; Hunt, D.E.; Junio, B.; Tyner, J.W.; Druker, B.J. Durable Disease Control with MEK Inhibition in a Patient with NRAS-mutated Atypical Chronic Myeloid Leukemia. Cureus 2015, 7, e414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesarwani, M.; Kincaid, Z.; Azam, M. MEK/ERK addiction in CNL/aCML. Oncotarget 2017, 8, 99215–99216. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, K.; Spitzer, B.; Kentsis, A. Therapeutic re-activation of protein phosphatase 2A in acute myeloid leukemia. Front. Oncol. 2015, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Coccaro, N.; Tota, G.; Zagaria, A.; Anelli, L.; Specchia, G.; Albano, F. SETBP1 dysregulation in congenital disorders and myeloid neoplasms. Oncotarget 2017, 8, 51920–51935. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Case 1 | Clinical Case 2 | |
|---|---|---|
| Age (years) | 41 | 71 |
| WBC | 32.12 × 109/L | 29.35 × 109/L |
| Differential (PB) | hypogranulated neutrophils 46% band-cells 12% eosinophils 2% basophils - monocytes 6% lymphocytes 14% myeloblasts - promyelocytes 2% metamyelocytes 12% myelocites 6% | neutrophils with P-H abnormality 30% band-cells 17% eosinophils - basophils - monocytes 15% lymphocytes 9% myeloblasts 1% promyelocytes 14% metamyelocytes 6% myelocites 8% |
| Hb | 13.5 g/dL | 14.9 g/dL |
| Plts | 103 × 109/L | 239 × 109/L |
| BM blasts | 2% | 4% |
| Screening for mutations in JAK2, CALR and MPL | negative | negative |
| BCR/ABL | negative | negative |
| Gene mutations identified by NGS panel | TET2
p.Q635* (VAF 37.09%) TET2 p.C1221Y (VAF 42.23%) EZH2 p.R690H (VAF 82.89%) | SETBP1 p.D868N (VAF 47.1%) SRSF2 p.P95H (VAF 52.34%) TET2 p.Y1245Lfs*22 (VAF 52.8%) |
| Screening for ETNK1 mutations | negative | ETNK1 p.H243P |
| Karyotype | 46, XY (20) | 46, XY (20) |
| Treatment | Decitabine 3cycles PR with residual thrombocytopenia HSCT | Peghilated IFN alpha PR |
| Follow-up time (months after diagnosis) | 8 months | 10 months |
| Status at last follow up | Alive in complete remission | Alive with stable disease and partial hematological response |
| WHO 2016 Diagnostic Criteria for aCML |
|---|
| Peripheral blood leukocytosis (WBC count ≥ 13 × 109/L) because of increased numbers of neutrophils and their precursors with prominent dysgranulopoiesis |
| Neutrophil precursors (promyelocytes, myelocytes, metamyelocytes) ≥ 10% of leukocytes |
| No Ph chromosome or BCR-ABL1 fusion gene and not meeting criteria for PV, ET, or PMF * |
| No evidence of PDGFRA, PDGFRB, FGFR1 rearrangement, or PCM1-JAK2 |
| Minimal absolute basophilia; basophils usually < 2% of leukocytes |
| No or minimal absolute monocytosis; monocytes usually < 10% of leukocytes |
| Hypercellular bone marrow with granulocytic proliferation and granulocytic dysplasia, with or without dysplasia in the erythroid and megakaryocytic lineages |
| Less than 20% blasts in the blood and bone marrow |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crisà, E.; Nicolosi, M.; Ferri, V.; Favini, C.; Gaidano, G.; Patriarca, A. Atypical Chronic Myeloid Leukemia: Where Are We Now? Int. J. Mol. Sci. 2020, 21, 6862. https://doi.org/10.3390/ijms21186862
Crisà E, Nicolosi M, Ferri V, Favini C, Gaidano G, Patriarca A. Atypical Chronic Myeloid Leukemia: Where Are We Now? International Journal of Molecular Sciences. 2020; 21(18):6862. https://doi.org/10.3390/ijms21186862
Chicago/Turabian StyleCrisà, Elena, Maura Nicolosi, Valentina Ferri, Chiara Favini, Gianluca Gaidano, and Andrea Patriarca. 2020. "Atypical Chronic Myeloid Leukemia: Where Are We Now?" International Journal of Molecular Sciences 21, no. 18: 6862. https://doi.org/10.3390/ijms21186862
APA StyleCrisà, E., Nicolosi, M., Ferri, V., Favini, C., Gaidano, G., & Patriarca, A. (2020). Atypical Chronic Myeloid Leukemia: Where Are We Now? International Journal of Molecular Sciences, 21(18), 6862. https://doi.org/10.3390/ijms21186862