Molecular Pathogenesis and Treatment Perspectives for Hypereosinophilia and Hypereosinophilic Syndromes

 , , ,
, , ,  and
and
Abstract
:1. Introduction
1.1. Eosinophil Development
1.2. Eosinophil Contents, Biology and Homeostatic Immune Role
1.3. Eosinophil Recruitment into Blood and Tissue, Survival and Death
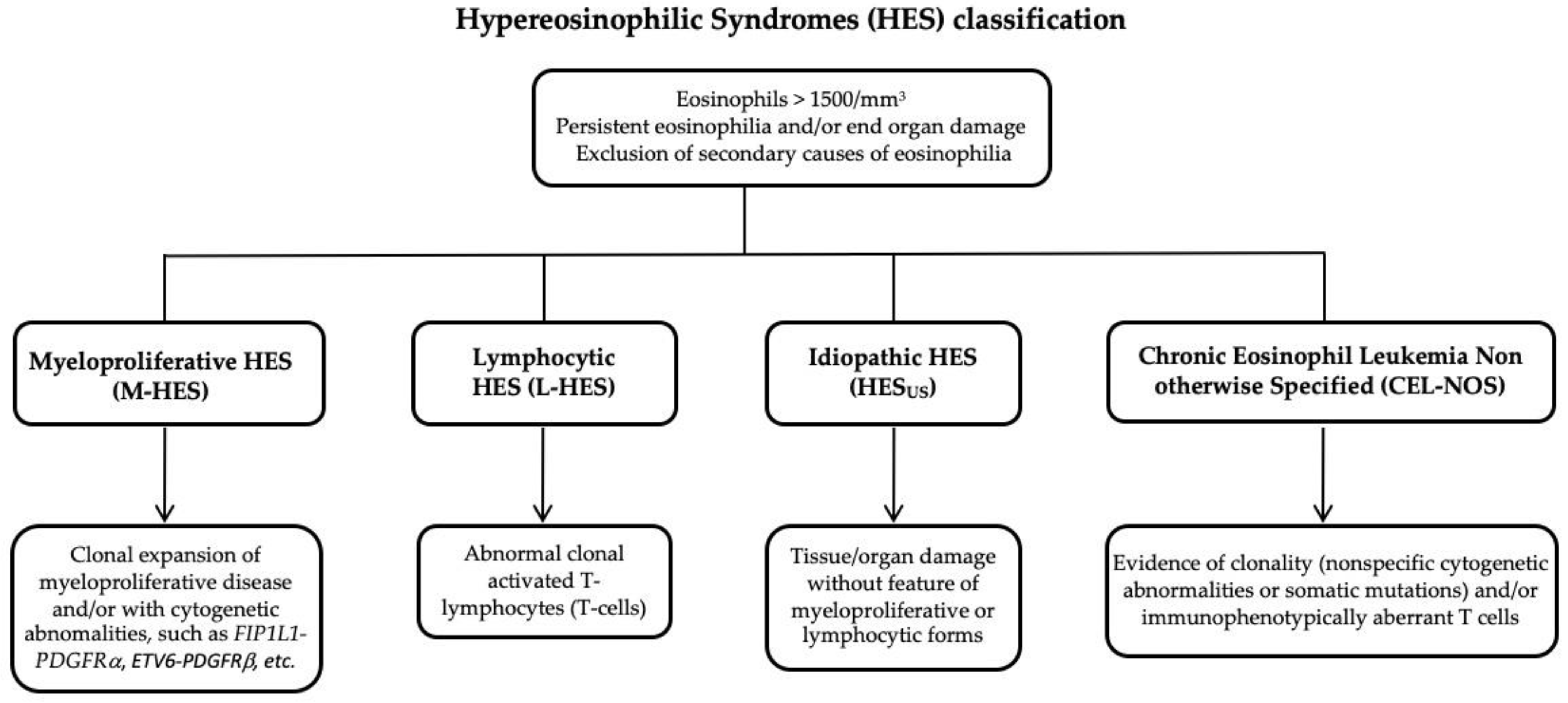
2. Classification of Hypereosinophilia and Hypereosinophilic Syndrome
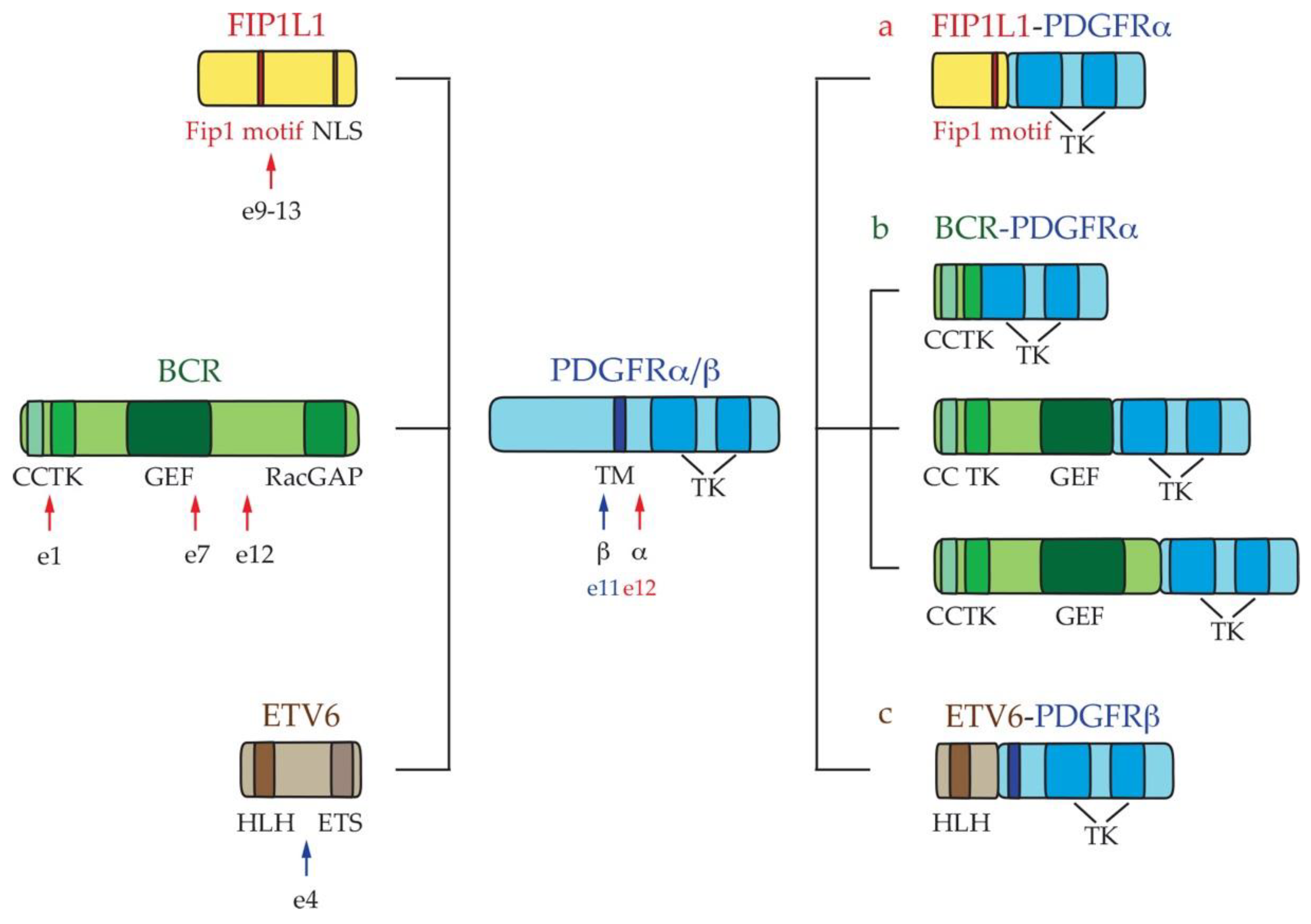
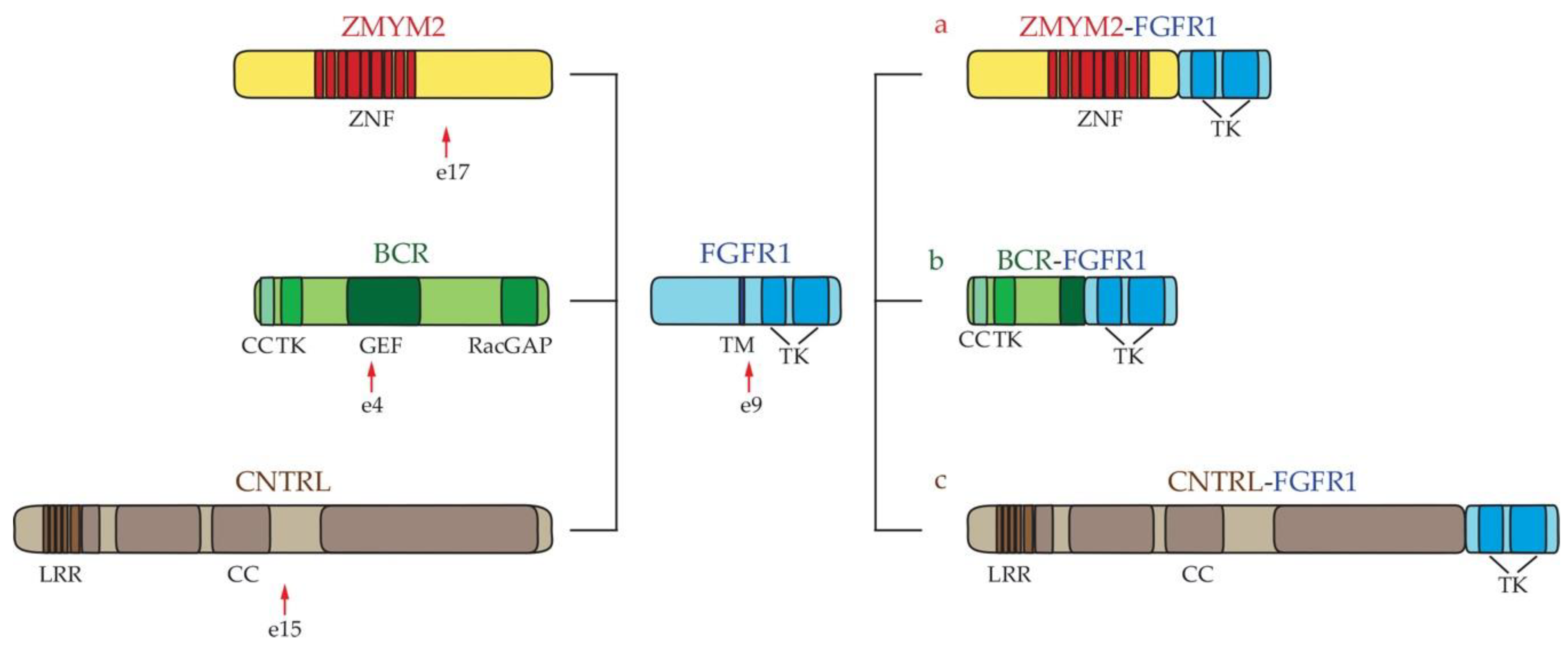
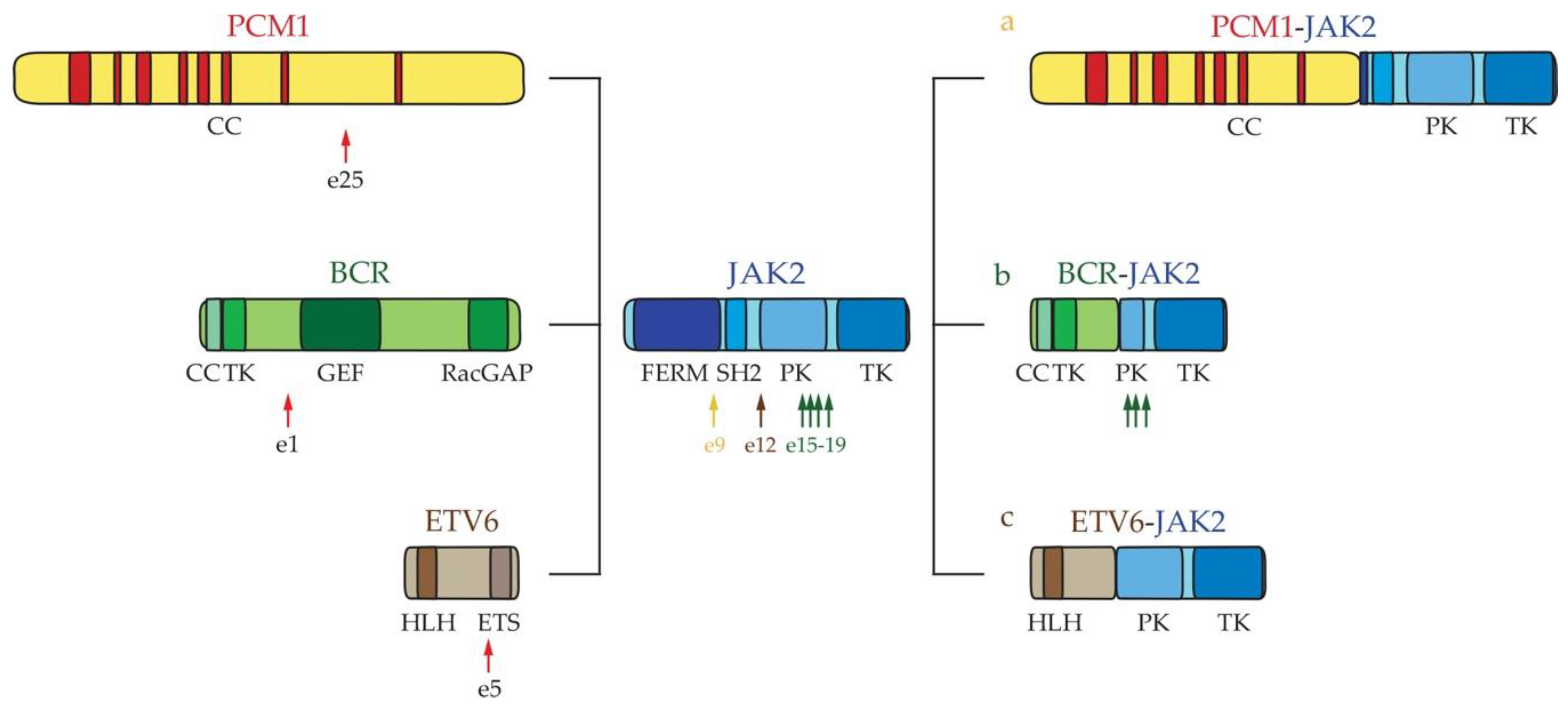
2.1. Myeloid Hypereosinophilic Syndrome
2.2. Lymphocytic Hypereosinophilic Syndrome
2.3. Idiopathic Hypereosinophilic Syndrome and Chronic Eosinophilic Leukemia non Otherwise Specified
3. Molecular Pathogenesis in Hypereosinophilic Syndrome
Next Generations Sequencing Approaches to Investigate DNA Mutations in Patients with Eosinophilic Disorders
4. Therapeutic Option for Patients with Eosinophilic Disorders
4.1. Corticosteroids
4.2. Cytotoxic Agents
4.3. Tyrosine Kinase Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABL1 | Abelson murine leukemia |
| AEC | Absolute Eosinophil Count |
| ALL | Acute Lymphoblastic Leukemia |
| BCR | Breakpoint cluster region |
| CEL-NOS | Chronic Eosinophilic Leukemia non otherwise specified |
| CML | Chronic Myeloid Leukemia |
| CMML | Chronic Myelomonocytic Leukemia |
| CNL | Chronic Neutrophilic Leukemia |
| CNTRL | Centriolin |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FIPL1 | FIP1-like 1 |
| FLT3 | fms-like tyrosine kinase 3 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| GUSβ | β-glucuronidase |
| HE | Hypereosinophilia |
| HES | Hypereosinophilia syndrome |
| IL-5 | Interleukin 5 |
| JAK2 | janus kinase 2 |
| MDS | myelodysplastic sindromes |
| MPN | myeloproliferative neoplasms |
| PDGFRα | Platelet-Derived Growth Factor Receptor alpha |
| PCM1 | Periocentriolar material 1 |
| TARC | Thymus and activation-regulated chemokine |
| TK | Tyrosine kinase |
| TKI | Tyrosine kinase inhibitor |
| TFR | Treatment-free remission |
| ZMYM2 | Zinc finger MYM-type protein 2 |
References
- Rothenberg, M.E. Eosinophilia. N. Engl. J. Med. 1998, 338, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Kovalszki, A.; Weller, P.F. Eosinophilia. Prim. Care 2016, 43, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, K.R.; Ackerman, S.J. Eosinophil granule proteins: Form and function. J. Biol. Chem. 2014, 289, 17406–17415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorak, A.M.; Letourneau, L.; Login, G.R.; Weller, P.F.; Ackerman, S.J. Ultrastructural localization of the Charcot-Leyden crystal protein (lysophospholipase) to a distinct crystalloid-free granule population in mature human eosinophils. Blood 1988, 72, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giembycz, M.A.; Lindsay, M.A. Pharmacology of the eosinophil. Pharmacol. Rev. 1999, 51, 213–340. [Google Scholar]
- Shamri, R.; Xenakis, J.J.; Spencer, L.A. Eosinophils in innate immunity: An evolving story. Cell Tissue Res. 2011, 343, 57–83. [Google Scholar] [CrossRef] [Green Version]
- Persson, T.; Andersson, P.; Bodelsson, M.; Laurell, M.; Malm, J.; Egesten, A. Bactericidal activity of human eosinophilic granulocytes against Escherichia coli. Infect. Immun. 2001, 69, 3591–3596. [Google Scholar] [CrossRef] [Green Version]
- Sabogal Pineros, Y.S.; Bal, S.M.; Dijkhuis, A.; Majoor, C.J.; Dierdorp, B.S.; Dekker, T.; Hoefsmit, E.P.; Bonta, P.I.; Picavet, D.; van der Wel, N.N.; et al. Eosinophils capture viruses, a capacity that is defective in asthma. Allergy 2019, 74, 1898–1909. [Google Scholar] [CrossRef]
- Weller, P.F.; Spencer, L.A. Functions of tissue-resident eosinophils. Nat. Rev. Immunol. 2017, 17, 746–760. [Google Scholar] [CrossRef]
- Goh, Y.P.; Henderson, N.C.; Heredia, J.E.; Red Eagle, A.; Odegaard, J.I.; Lehwald, N.; Nguyen, K.D.; Sheppard, D.; Mukundan, L.; Locksley, R.M.; et al. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9914–9919. [Google Scholar] [CrossRef] [Green Version]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeziorska, M.; Salamonsen, L.A.; Woolley, D.E. Mast cell and eosinophil distribution and activation in human endometrium throughout the menstrual cycle. Biol. Reprod. 1995, 53, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.S.; Won, T.; Hou, X.; Chen, G.; Bracamonte-Baran, W.; Talor, M.V.; Jurcova, I.; Szarszoi, O.; Curnova, L.; Striz, I.; et al. Innate Lymphoid Cells Play a Pathogenic Role in Pericarditis. Cell Rep. 2020, 30, 2989–3003.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, K.; Malmhall, C.; Ramos-Ramirez, P.; Radinger, M. Bone marrow type 2 innate lymphoid cells: A local source of interleukin-5 in interleukin-33-driven eosinophilia. Immunology 2018, 153, 268–278. [Google Scholar] [CrossRef]
- Roufosse, F. Targeting the Interleukin-5 Pathway for Treatment of Eosinophilic Conditions Other than Asthma. Front. Med. 2018, 5, 49. [Google Scholar] [CrossRef] [Green Version]
- Dent, L.A.; Strath, M.; Mellor, A.L.; Sanderson, C.J. Eosinophilia in transgenic mice expressing interleukin 5. J. Exp. Med. 1990, 172, 1425–1431. [Google Scholar] [CrossRef] [Green Version]
- Esnault, S.; Kelly, E.A. Essential Mechanisms of Differential Activation of Eosinophils by IL-3 Compared to GM-CSF and IL-5. Crit. Rev. Immunol. 2016, 36, 429–444. [Google Scholar] [CrossRef] [Green Version]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef]
- McBrien, C.N.; Menzies-Gow, A. The Biology of Eosinophils and Their Role in Asthma. Front. Med. 2017, 4, 93. [Google Scholar] [CrossRef]
- O’Sullivan, J.A.; Carroll, D.J.; Bochner, B.S. Glycobiology of Eosinophilic Inflammation: Contributions of Siglecs, Glycans, and Other Glycan-Binding Proteins. Front. Med. 2017, 4, 116. [Google Scholar] [CrossRef] [Green Version]
- Legrand, F.; Cao, Y.; Wechsler, J.B.; Zhu, X.; Zimmermann, N.; Rampertaap, S.; Monsale, J.; Romito, K.; Youngblood, B.A.; Brock, E.C.; et al. Sialic acid-binding immunoglobulin-like lectin (Siglec) 8 in patients with eosinophilic disorders: Receptor expression and targeting using chimeric antibodies. J. Allergy Clin. Immunol. 2019, 143, 2227–2237. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Klion, A.D.; Horny, H.P.; Roufosse, F.; Gotlib, J.; Weller, P.F.; Hellmann, A.; Metzgeroth, G.; Leiferman, K.M.; Arock, M.; et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J. Allergy Clin. Immunol. 2012, 130, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Shomali, W.; Gotlib, J. World Health Organization-defined eosinophilic disorders: 2019 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2019, 94, 1149–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattis, D.M.; Wang, S.A.; Lu, C.M. Contemporary Classification and Diagnostic Evaluation of Hypereosinophilia. Am. J. Clin. Pathol. 2020. [Google Scholar] [CrossRef]
- Schwaab, J.; Jawhar, M.; Naumann, N.; Schmitt-Graeff, A.; Fabarius, A.; Horny, H.P.; Cross, N.C.; Hofmann, W.K.; Reiter, A.; Metzgeroth, G. Diagnostic challenges in the work up of hypereosinophilia: Pitfalls in bone marrow core biopsy interpretation. Ann. Hematol. 2016, 95, 557–562. [Google Scholar] [CrossRef]
- Ackerman, S.J.; Bochner, B.S. Mechanisms of eosinophilia in the pathogenesis of hypereosinophilic disorders. Immunol. Allergy Clin. N. Am. 2007, 27, 357–375. [Google Scholar] [CrossRef] [Green Version]
- Lefevre, G.; Copin, M.C.; Staumont-Salle, D.; Avenel-Audran, M.; Aubert, H.; Taieb, A.; Salles, G.; Maisonneuve, H.; Ghomari, K.; Ackerman, F.; et al. The lymphoid variant of hypereosinophilic syndrome: Study of 21 patients with CD3-CD4+ aberrant T-cell phenotype. Medicine 2014, 93, 255–266. [Google Scholar] [CrossRef]
- Leru, P.M. Eosinophilic disorders: Evaluation of current classification and diagnostic criteria, proposal of a practical diagnostic algorithm. Clin. Transl. Allergy 2019, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Weller, P.F.; Bubley, G.J. The idiopathic hypereosinophilic syndrome. Blood 1994, 83, 2759–2779. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. In WHO Classification of Tumours, Revised, 4th ed.; IARC Pubblication: Lyon, France, 2017; Volume 2. [Google Scholar]
- Dispenza, M.C.; Bochner, B.S. Diagnosis and Novel Approaches to the Treatment of Hypereosinophilic Syndromes. Curr. Hematol. Malig. Rep. 2018, 13, 191–201. [Google Scholar] [CrossRef]
- Simon, H.U.; Rothenberg, M.E.; Bochner, B.S.; Weller, P.F.; Wardlaw, A.J.; Wechsler, M.E.; Rosenwasser, L.J.; Roufosse, F.; Gleich, G.J.; Klion, A.D. Refining the definition of hypereosinophilic syndrome. J. Allergy Clin. Immunol. 2010, 126, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klion, A.D. Eosinophilic myeloproliferative disorders. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havelange, V.; Demoulin, J.B. Review of current classification, molecular alterations, and tyrosine kinase inhibitor therapies in myeloproliferative disorders with hypereosinophilia. J. Blood Med. 2013, 4, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, A.; Gotlib, J. Myeloid neoplasms with eosinophilia. Blood 2017, 129, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Roufosse, F.; Cogan, E.; Goldman, M. Recent advances in pathogenesis and management of hypereosinophilic syndromes. Allergy 2004, 59, 673–689. [Google Scholar] [CrossRef]
- Roufosse, F. Hypereosinophilic syndrome variants: Diagnostic and therapeutic considerations. Haematologica 2009, 94, 1188–1193. [Google Scholar] [CrossRef] [Green Version]
- de Lavareille, A.; Roufosse, F.; Schmid-Grendelmeier, P.; Roumier, A.S.; Schandene, L.; Cogan, E.; Simon, H.U.; Goldman, M. High serum thymus and activation-regulated chemokine levels in the lymphocytic variant of the hypereosinophilic syndrome. J. Allergy Clin. Immunol. 2002, 110, 476–479. [Google Scholar] [CrossRef]
- Szuber, N.; Elliott, M.; Tefferi, A. Chronic neutrophilic leukemia: 2020 update on diagnosis, molecular genetics, prognosis, and management. Am. J. Hematol. 2020, 95, 212–224. [Google Scholar] [CrossRef]
- Solary, E.; Itzykson, R. How I treat chronic myelomonocytic leukemia. Blood 2017, 130, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Stagno, F.; Vigneri, P.; Del Fabro, V.; Stella, S.; Cupri, A.; Massimino, M.; Consoli, C.; Tambe, L.; Consoli, M.L.; Antolino, A.; et al. Influence of complex variant chromosomal translocations in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Acta Oncol. 2010, 49, 506–508. [Google Scholar] [CrossRef]
- Vigneri, P.; Stagno, F.; Stella, S.; Cupri, A.; Forte, S.; Massimino, M.; Antolino, A.; Siragusa, S.; Mannina, D.; Impera, S.S.; et al. High BCR-ABL/GUS(IS) Levels at Diagnosis of Chronic Phase CML Are Associated with Unfavorable Responses to Standard-Dose Imatinib. Clin. Cancer Res. 2017, 23, 7189–7198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.A. The Diagnostic Work-Up of Hypereosinophilia. Pathobiology 2019, 86, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Reilly, J.T. Cytogenetic and molecular genetic aspects of idiopathic myelofibrosis. Acta Haematol. 2002, 108, 113–119. [Google Scholar] [CrossRef]
- Yarden, Y.; Escobedo, J.A.; Kuang, W.J.; Yang-Feng, T.L.; Daniel, T.O.; Tremble, P.M.; Chen, E.Y.; Ando, M.E.; Harkins, R.N.; Francke, U.; et al. Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature 1986, 323, 226–232. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. Platelet-derived growth factor receptor signals. J. Biol. Chem. 1994, 269, 32023–32026. [Google Scholar] [CrossRef]
- Heldin, C.H.; Ostman, A.; Ronnstrand, L. Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta 1998, 1378, F79–F113. [Google Scholar] [CrossRef]
- Valgeirsdottir, S.; Paukku, K.; Silvennoinen, O.; Heldin, C.H.; Claesson-Welsh, L. Activation of Stat5 by platelet-derived growth factor (PDGF) is dependent on phosphorylation sites in PDGF beta-receptor juxtamembrane and kinase insert domains. Oncogene 1998, 16, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Sachsenmaier, C.; Sadowski, H.B.; Cooper, J.A. STAT activation by the PDGF receptor requires juxtamembrane phosphorylation sites but not Src tyrosine kinase activation. Oncogene 1999, 18, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Robyn, J.; Lemery, S.; McCoy, J.P.; Kubofcik, J.; Kim, Y.J.; Pack, S.; Nutman, T.B.; Dunbar, C.; Klion, A.D. Multilineage involvement of the fusion gene in patients with FIP1L1/PDGFRA-positive hypereosinophilic syndrome. Br. J. Haematol. 2006, 132, 286–292. [Google Scholar] [CrossRef]
- Cools, J.; DeAngelo, D.J.; Gotlib, J.; Stover, E.H.; Legare, R.D.; Cortes, J.; Kutok, J.; Clark, J.; Galinsky, I.; Griffin, J.D.; et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N. Engl. J. Med. 2003, 348, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Elling, C.; Erben, P.; Walz, C.; Frickenhaus, M.; Schemionek, M.; Stehling, M.; Serve, H.; Cross, N.C.; Hochhaus, A.; Hofmann, W.K.; et al. Novel imatinib-sensitive PDGFRA-activating point mutations in hypereosinophilic syndrome induce growth factor independence and leukemia-like disease. Blood 2011, 117, 2935–2943. [Google Scholar] [CrossRef] [PubMed]
- Maric, I.; Robyn, J.; Metcalfe, D.D.; Fay, M.P.; Carter, M.; Wilson, T.; Fu, W.; Stoddard, J.; Scott, L.; Hartsell, M.; et al. KIT D816V-associated systemic mastocytosis with eosinophilia and FIP1L1/PDGFRA-associated chronic eosinophilic leukemia are distinct entities. J. Allergy Clin. Immunol. 2007, 120, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J.; Cools, J. Five years since the discovery of FIP1L1-PDGFRA: What we have learned about the fusion and other molecularly defined eosinophilias. Leukemia 2008, 22, 1999–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotlib, J.; Cross, N.C.; Gilliland, D.G. Eosinophilic disorders: Molecular pathogenesis, new classification, and modern therapy. Best Pract. Res. Clin. Haematol. 2006, 19, 535–569. [Google Scholar] [CrossRef]
- Reiter, A.; Walz, C.; Watmore, A.; Schoch, C.; Blau, I.; Schlegelberger, B.; Berger, U.; Telford, N.; Aruliah, S.; Yin, J.A.; et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005, 65, 2662–2667. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Sydney Sir Philip, J.K.; Weinberg, O.; Tam, W.; Sadigh, S.; Lake, J.I.; Margolskee, E.M.; Rogers, H.J.; Miranda, R.N.; Bueso-Ramos, C.C.; et al. Hematopoietic neoplasms with 9p24/JAK2 rearrangement: A multicenter study. Mod. Pathol. 2019, 32, 490–498. [Google Scholar] [CrossRef]
- Thakral, B.; Muzzafar, T.; Wang, S.A.; Medeiros, L.J. Myeloid neoplasm with eosinophilia and BCR-JAK2/t(9;22)(p24;q11.2) morphologically mimicking chronic myeloid leukemia. Ann. Diagn. Pathol. 2020, 44, 151405. [Google Scholar] [CrossRef]
- Yu, J.; Deuel, T.F.; Kim, H.R. Platelet-derived growth factor (PDGF) receptor-alpha activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-beta -induced phenotypic transformation. J. Biol. Chem. 2000, 275, 19076–19082. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, K.; Matsumura, I.; Ezoe, S.; Tokunaga, M.; Yasumi, M.; Satoh, Y.; Shibayama, H.; Tanaka, H.; Iwama, A.; Kanakura, Y. FIP1L1-PDGFRalpha imposes eosinophil lineage commitment on hematopoietic stem/progenitor cells. J. Biol. Chem. 2009, 284, 7719–7732. [Google Scholar] [CrossRef] [Green Version]
- Buffa, P.; Romano, C.; Pandini, A.; Massimino, M.; Tirro, E.; Di Raimondo, F.; Manzella, L.; Fraternali, F.; Vigneri, P.G. BCR-ABL residues interacting with ponatinib are critical to preserve the tumorigenic potential of the oncoprotein. FASEB J. 2014, 28, 1221–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Hochhaus, A.; Bolufer, P.; Reiter, A.; Fernandez, J.M.; Senent, L.; Cervera, J.; Moscardo, F.; Sanz, M.A.; Cross, N.C. The t(4;22)(q12;q11) in atypical chronic myeloid leukaemia fuses BCR to PDGFRA. Hum. Mol. Genet. 2002, 11, 1391–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trempat, P.; Villalva, C.; Laurent, G.; Armstrong, F.; Delsol, G.; Dastugue, N.; Brousset, P. Chronic myeloproliferative disorders with rearrangement of the platelet-derived growth factor alpha receptor: A new clinical target for STI571/Glivec. Oncogene 2003, 22, 5702–5706. [Google Scholar] [CrossRef] [Green Version]
- Safley, A.M.; Sebastian, S.; Collins, T.S.; Tirado, C.A.; Stenzel, T.T.; Gong, J.Z.; Goodman, B.K. Molecular and cytogenetic characterization of a novel translocation t(4;22) involving the breakpoint cluster region and platelet-derived growth factor receptor-alpha genes in a patient with atypical chronic myeloid leukemia. Genes Chromosomes Cancer 2004, 40, 44–50. [Google Scholar] [CrossRef]
- Keene, P.; Mendelow, B.; Pinto, M.R.; Bezwoda, W.; MacDougall, L.; Falkson, G.; Ruff, P.; Bernstein, R. Abnormalities of chromosome 12p13 and malignant proliferation of eosinophils: A nonrandom association. Br. J. Haematol. 1987, 67, 25–31. [Google Scholar] [CrossRef]
- Curtis, C.E.; Grand, F.H.; Waghorn, K.; Sahoo, T.P.; George, J.; Cross, N.C. A novel ETV6-PDGFRB fusion transcript missed by standard screening in a patient with an imatinib responsive chronic myeloproliferative disease. Leukemia 2007, 21, 1839–1841. [Google Scholar] [CrossRef]
- Toffalini, F.; Hellberg, C.; Demoulin, J.B. Critical role of the platelet-derived growth factor receptor (PDGFR) beta transmembrane domain in the TEL-PDGFRbeta cytosolic oncoprotein. J. Biol. Chem. 2010, 285, 12268–12278. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.A.; Hasserjian, R.P.; Tam, W.; Tsai, A.G.; Geyer, J.T.; George, T.I.; Foucar, K.; Rogers, H.J.; Hsi, E.D.; Rea, B.A.; et al. Bone marrow morphology is a strong discriminator between chronic eosinophilic leukemia, not otherwise specified and reactive idiopathic hypereosinophilic syndrome. Haematologica 2017, 102, 1352–1360. [Google Scholar] [CrossRef] [Green Version]
- Montano-Almendras, C.P.; Essaghir, A.; Schoemans, H.; Varis, I.; Noel, L.A.; Velghe, A.I.; Latinne, D.; Knoops, L.; Demoulin, J.B. ETV6-PDGFRB and FIP1L1-PDGFRA stimulate human hematopoietic progenitor cell proliferation and differentiation into eosinophils: The role of nuclear factor-kappaB. Haematologica 2012, 97, 1064–1072. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iurlo, A.; Cattaneo, D.; Gianelli, U. Hypereosinophilic syndromes in the precision medicine era: Clinical, molecular aspects and therapeutic approaches (targeted therapies). Expert Rev. Hematol. 2019, 12, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Peiris, M.N.; Meyer, A.N.; Nelson, K.N.; Bisom-Rapp, E.W.; Donoghue, D.J. Oncogenic fusion protein BCR-FGFR1 requires the breakpoint cluster region-mediated oligomerization and chaperonin Hsp90 for activation. Haematologica 2020, 105, 1262–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.; Reiter, A.; Smedley, D.; Goldman, J.M.; Cross, N.C. The genomic structure of ZNF198 and location of breakpoints in the t(8;13) myeloproliferative syndrome. Genomics 1999, 55, 118–121. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Elli, E.M.; Barate, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [Green Version]
- Bain, B.J.; Fletcher, S.H. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol. Allergy Clin. N. Am. 2007, 27, 377–388. [Google Scholar] [CrossRef]
- Stewart, K.; Carstairs, K.C.; Dube, I.D.; Keating, A. Neutrophilic myelofibrosis presenting as Philadelphia chromosome negative BCR non-rearranged chronic myeloid leukemia. Am. J. Hematol. 1990, 34, 59–63. [Google Scholar] [CrossRef]
- Elnaggar, M.M.; Agersborg, S.; Sahoo, T.; Girgin, A.; Ma, W.; Rakkhit, R.; Zorrilla, I.; Leal, A. BCR-JAK2 fusion as a result of a translocation (9;22)(p24;q11.2) in a patient with CML-like myeloproliferative disease. Mol. Cytogenet. 2012, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.R.; Rogers, H.J.; Chandra, P.K.; Prescott, J.L.; Mukherjee, S. Myeloid neoplasm with eosinophilia and ETV6-JAK2 fusion. Leuk. Lymphoma 2020, 61, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Griesinger, F.; Hennig, H.; Hillmer, F.; Podleschny, M.; Steffens, R.; Pies, A.; Wormann, B.; Haase, D.; Bohlander, S.K. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11.2) translocation in a patient with a clinically typical chronic myeloid leukemia. Genes Chromosomes Cancer 2005, 44, 329–333. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Greipp, P.T.; Rangan, A.; Mai, M.; Chen, D.; Reichard, K.K.; Nelsen, L.L.; Pardanani, A.; Hanson, C.A.; Viswanatha, D.S. BCR-JAK2 fusion in a myeloproliferative neoplasm with associated eosinophilia. Cancer Genet. 2016, 209, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Peeters, P.; Raynaud, S.D.; Cools, J.; Wlodarska, I.; Grosgeorge, J.; Philip, P.; Monpoux, F.; Van Rompaey, L.; Baens, M.; Van den Berghe, H.; et al. Fusion of TEL, the ETS-variant gene 6 (ETV6), to the receptor-associated kinase JAK2 as a result of t(9;12) in a lymphoid and t(9;15;12) in a myeloid leukemia. Blood 1997, 90, 2535–2540. [Google Scholar] [CrossRef] [Green Version]
- Bain, B.J.; Ahmad, S. Should myeloid and lymphoid neoplasms with PCM1-JAK2 and other rearrangements of JAK2 be recognized as specific entities? Br. J. Haematol. 2014, 166, 809–817. [Google Scholar] [CrossRef]
- Cirmena, G.; Aliano, S.; Fugazza, G.; Bruzzone, R.; Garuti, A.; Bocciardi, R.; Bacigalupo, A.; Ravazzolo, R.; Ballestrero, A.; Sessarego, M. A BCR-JAK2 fusion gene as the result of a t(9;22)(p24;q11) in a patient with acute myeloid leukemia. Cancer Genet. Cytogenet. 2008, 183, 105–108. [Google Scholar] [CrossRef]
- Patterer, V.; Schnittger, S.; Kern, W.; Haferlach, T.; Haferlach, C. Hematologic malignancies with PCM1-JAK2 gene fusion share characteristics with myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, and FGFR1. Ann. Hematol. 2013, 92, 759–769. [Google Scholar] [CrossRef]
- Schwaller, J. Modeling ETV6-JAK2-induced leukemia: Insights from the zebrafish. Haematologica 2012, 97, 1783–1785. [Google Scholar] [CrossRef]
- Cuesta-Dominguez, A.; Ortega, M.; Ormazabal, C.; Santos-Roncero, M.; Galan-Diez, M.; Steegmann, J.L.; Figuera, A.; Arranz, E.; Vizmanos, J.L.; Bueren, J.A.; et al. Transforming and tumorigenic activity of JAK2 by fusion to BCR: Molecular mechanisms of action of a novel BCR-JAK2 tyrosine-kinase. PLoS ONE 2012, 7, e32451. [Google Scholar] [CrossRef] [Green Version]
- Falchi, L.; Mehrotra, M.; Newberry, K.J.; Lyle, L.M.; Lu, G.; Patel, K.P.; Luthra, R.; Popat, U.; Verstovsek, S. ETV6-FLT3 fusion gene-positive, eosinophilia-associated myeloproliferative neoplasm successfully treated with sorafenib and allogeneic stem cell transplant. Leukemia 2014, 28, 2090–2092. [Google Scholar] [CrossRef] [Green Version]
- Chonabayashi, K.; Hishizawa, M.; Matsui, M.; Kondo, T.; Ohno, T.; Ishikawa, T.; Takaori-Kondo, A. Successful allogeneic stem cell transplantation with long-term remission of ETV6/FLT3-positive myeloid/lymphoid neoplasm with eosinophilia. Ann. Hematol. 2014, 93, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.E.; Groh, M.; Lefevre, G. (A Critical Appraisal of) Classification of Hypereosinophilic Disorders. Front. Med. 2017, 4, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maric, I.; Sun, X. Advances in diagnosis of mastocytosis and hypereosinophilic syndrome. Semin. Hematol. 2019, 56, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Helbig, G.; Wieczorkiewicz, A.; Dziaczkowska-Suszek, J.; Majewski, M.; Kyrcz-Krzemien, S. T-cell abnormalities are present at high frequencies in patients with hypereosinophilic syndrome. Haematologica 2009, 94, 1236–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Hussaini, M.; Zhang, H.; Shao, H.; Qin, D.; Zhang, X.; Ma, Z.; Hussnain Naqvi, S.M.; Zhang, L.; Moscinski, L.C. Comparison of the Mutational Profiles of Primary Myelofibrosis, Polycythemia Vera, and Essential Thrombocytosis. Am. J. Clin. Pathol. 2017, 147, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Hussaini, M.O.; Mirza, A.S.; Komrokji, R.; Lancet, J.; Padron, E.; Song, J. Genetic Landscape of Acute Myeloid Leukemia Interrogated by Next-generation Sequencing: A Large Cancer Center Experience. Cancer Genom. Proteom. 2018, 15, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef] [Green Version]
- Soverini, S.; Bavaro, L.; De Benedittis, C.; Martelli, M.; Iurlo, A.; Orofino, N.; Sica, S.; Sora, F.; Lunghi, F.; Ciceri, F.; et al. Prospective assessment of NGS-detectable mutations in CML patients with nonoptimal response: The NEXT-in-CML study. Blood 2020, 135, 534–541. [Google Scholar] [CrossRef]
- Soverini, S.; Abruzzese, E.; Bocchia, M.; Bonifacio, M.; Galimberti, S.; Gozzini, A.; Iurlo, A.; Luciano, L.; Pregno, P.; Rosti, G.; et al. Next-generation sequencing for BCR-ABL1 kinase domain mutation testing in patients with chronic myeloid leukemia: A position paper. J. Hematol. Oncol. 2019, 12, 131. [Google Scholar] [CrossRef] [Green Version]
- Baer, C.; Muehlbacher, V.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular genetic characterization of myeloid/lymphoid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2. Haematologica 2018, 103, e348–e350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardanani, A.; Lasho, T.; Wassie, E.; Finke, C.; Zblewski, D.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Predictors of survival in WHO-defined hypereosinophilic syndrome and idiopathic hypereosinophilia and the role of next-generation sequencing. Leukemia 2016, 30, 1924–1926. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.A.; Tam, W.; Tsai, A.G.; Arber, D.A.; Hasserjian, R.P.; Geyer, J.T.; George, T.I.; Czuchlewski, D.R.; Foucar, K.; Rogers, H.J.; et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod. Pathol. 2016, 29, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Seo, H.; Im, K.; Park, S.N.; Kim, S.M.; Lee, E.K.; Kim, J.A.; Lee, J.H.; Kwon, S.; Kim, M.; et al. Idiopathic hypereosinophilia is clonal disorder? Clonality identified by targeted sequencing. PLoS ONE 2017, 12, e0185602. [Google Scholar] [CrossRef] [Green Version]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lubke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2019, 33, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Ogbogu, P.U.; Bochner, B.S.; Butterfield, J.H.; Gleich, G.J.; Huss-Marp, J.; Kahn, J.E.; Leiferman, K.M.; Nutman, T.B.; Pfab, F.; Ring, J.; et al. Hypereosinophilic syndrome: A multicenter, retrospective analysis of clinical characteristics and response to therapy. J. Allergy Clin. Immunol. 2009, 124, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Helbig, G.; Wisniewska-Piaty, K.; Francuz, T.; Dziaczkowska-Suszek, J.; Kyrcz-Krzemien, S. Diversity of clinical manifestations and response to corticosteroids for idiopathic hypereosinophilic syndrome: Retrospective study in 33 patients. Leuk. Lymphoma 2013, 54, 807–811. [Google Scholar] [CrossRef]
- Fauci, A.S.; Harley, J.B.; Roberts, W.C.; Ferrans, V.J.; Gralnick, H.R.; Bjornson, B.H. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann. Intern. Med. 1982, 97, 78–92. [Google Scholar] [CrossRef]
- Ceretelli, S.; Capochiani, E.; Petrini, M. Interferon-alpha in the idiopathic hypereosinophilic syndrome: Consideration of five cases. Ann. Hematol. 1998, 77, 161–164. [Google Scholar] [CrossRef]
- Butterfield, J.H. Interferon treatment for hypereosinophilic syndromes and systemic mastocytosis. Acta Haematol. 2005, 114, 26–40. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.; Cortes, J.; Thomas, D.; Garcia-Manero, G.; Ferrajoli, A.; Faderl, S.; Richie, M.A.; Beran, M.; Giles, F.; et al. PEG-IFN-alpha-2b therapy in BCR-ABL-negative myeloproliferative disorders: Final result of a phase 2 study. Cancer 2007, 110, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Panch, S.R.; Bozik, M.E.; Brown, T.; Makiya, M.; Prussin, C.; Archibald, D.G.; Hebrank, G.T.; Sullivan, M.; Sun, X.; Wetzler, L.; et al. Dexpramipexole as an oral steroid-sparing agent in hypereosinophilic syndromes. Blood 2018, 132, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Stella, S.; Tirro, E.; Romano, C.; Pennisi, M.S.; Puma, A.; Manzella, L.; Zanghi, A.; Stagno, F.; Di Raimondo, F.; et al. Non ABL-directed inhibitors as alternative treatment strategies for chronic myeloid leukemia. Mol. Cancer 2018, 17, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerweel, P.E.; Te Boekhorst, P.A.W.; Levin, M.D.; Cornelissen, J.J. New Approaches and Treatment Combinations for the Management of Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 665. [Google Scholar] [CrossRef]
- Stagno, F.; Stella, S.; Spitaleri, A.; Pennisi, M.S.; Di Raimondo, F.; Vigneri, P. Imatinib mesylate in chronic myeloid leukemia: Frontline treatment and long-term outcomes. Expert Rev. Anticancer Ther. 2016, 16, 273–278. [Google Scholar] [CrossRef]
- Helbig, G. Imatinib for the treatment of hypereosinophilic syndromes. Expert Rev. Clin. Immunol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Wang, S.; Wilkes, M.C.; Leof, E.B.; Hirschberg, R. Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bose, P.; Masarova, L.; Verstovsek, S. Novel treatment strategies for myeloproliferative neoplasms. Rinsho Ketsueki 2019, 60, 1176–1185. [Google Scholar] [CrossRef]
- Rumi, E.; Milosevic, J.D.; Selleslag, D.; Casetti, I.; Lierman, E.; Pietra, D.; Cavalloni, C.; Bellini, M.; Milanesi, C.; Dambruoso, I.; et al. Efficacy of ruxolitinib in myeloid neoplasms with PCM1-JAK2 fusion gene. Ann. Hematol. 2015, 94, 1927–1928. [Google Scholar] [CrossRef]
- Schwaab, J.; Knut, M.; Haferlach, C.; Metzgeroth, G.; Horny, H.P.; Chase, A.; Tapper, W.; Score, J.; Waghorn, K.; Naumann, N.; et al. Limited duration of complete remission on ruxolitinib in myeloid neoplasms with PCM1-JAK2 and BCR-JAK2 fusion genes. Ann. Hematol. 2015, 94, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.; Erben, P.; Ritter, M.; Bloor, A.; Metzgeroth, G.; Telford, N.; Haferlach, C.; Haferlach, T.; Gesk, S.; Score, J.; et al. Response of ETV6-FLT3-positive myeloid/lymphoid neoplasm with eosinophilia to inhibitors of FMS-like tyrosine kinase 3. Blood 2011, 118, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Lierman, E.; Folens, C.; Stover, E.H.; Mentens, N.; Van Miegroet, H.; Scheers, W.; Boogaerts, M.; Vandenberghe, P.; Marynen, P.; Cools, J. Sorafenib is a potent inhibitor of FIP1L1-PDGFRalpha and the imatinib-resistant FIP1L1-PDGFRalpha T674I mutant. Blood 2006, 108, 1374–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.K.; Cook, R.M.; Zia-Amirhosseini, P.; Minthorn, E.; Sellers, T.S.; Maleeff, B.E.; Eustis, S.; Schwartz, L.W.; Tsui, P.; Appelbaum, E.R.; et al. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J. Allergy Clin. Immunol. 2001, 108, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.A.; Esnault, S.; Liu, L.Y.; Evans, M.D.; Johansson, M.W.; Mathur, S.; Mosher, D.F.; Denlinger, L.C.; Jarjour, N.N. Mepolizumab Attenuates Airway Eosinophil Numbers, but Not Their Functional Phenotype, in Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 1385–1395. [Google Scholar] [CrossRef]
- Busse, W.W.; Ring, J.; Huss-Marp, J.; Kahn, J.E. A review of treatment with mepolizumab, an anti-IL-5 mAb, in hypereosinophilic syndromes and asthma. J. Allergy Clin. Immunol. 2010, 125, 803–813. [Google Scholar] [CrossRef]
- Walsh, G.M. Reslizumab, a humanized anti-IL-5 mAb for the treatment of eosinophil-mediated inflammatory conditions. Curr. Opin. Mol. Ther. 2009, 11, 329–336. [Google Scholar]
- Bleecker, E.R.; FitzGerald, J.M.; Chanez, P.; Papi, A.; Weinstein, S.F.; Barker, P.; Sproule, S.; Gilmartin, G.; Aurivillius, M.; Werkstrom, V.; et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): A randomised, multicentre, placebo-controlled phase 3 trial. Lancet 2016, 388, 2115–2127. [Google Scholar] [CrossRef]
- Kuang, F.L.; Legrand, F.; Makiya, M.; Ware, J.; Wetzler, L.; Brown, T.; Magee, T.; Piligian, B.; Yoon, P.; Ellis, J.H.; et al. Benralizumab for PDGFRA-Negative Hypereosinophilic Syndrome. N. Engl. J. Med. 2019, 380, 1336–1346. [Google Scholar] [CrossRef]
- Kolbeck, R.; Kozhich, A.; Koike, M.; Peng, L.; Andersson, C.K.; Damschroder, M.M.; Reed, J.L.; Woods, R.; Dall’acqua, W.W.; Stephens, G.L.; et al. MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J. Allergy Clin. Immunol. 2010, 125, 1344–1353. [Google Scholar] [CrossRef]
- Massanari, M.; Holgate, S.T.; Busse, W.W.; Jimenez, P.; Kianifard, F.; Zeldin, R. Effect of omalizumab on peripheral blood eosinophilia in allergic asthma. Respir. Med. 2010, 104, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitini, V.; Teti, D.; Arrigo, C.; Righi, M. Alemtuzumab therapy for refractory idiopathic hypereosinophilic syndrome with abnormal T cells: A case report. Br. J. Haematol. 2004, 127, 477. [Google Scholar] [CrossRef] [PubMed]
- Sefcick, A.; Sowter, D.; DasGupta, E.; Russell, N.H.; Byrne, J.L. Alemtuzumab therapy for refractory idiopathic hypereosinophilic syndrome. Br. J. Haematol. 2004, 124, 558–559. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Tefferi, A.; Kantarjian, H.; Manshouri, T.; Luthra, R.; Pardanani, A.; Quintas-Cardama, A.; Ravandi, F.; Ault, P.; Bueso-Ramos, C.; et al. Alemtuzumab therapy for hypereosinophilic syndrome and chronic eosinophilic leukemia. Clin. Cancer Res. 2009, 15, 368–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roufosse, F. Management of Hypereosinophilic Syndromes. Immunol. Allergy Clin. N. Am. 2015, 35, 561–575. [Google Scholar] [CrossRef]
- Gotlib, J. World Health Organization-defined eosinophilic disorders: 2017 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2017, 92, 1243–1259. [Google Scholar] [CrossRef] [Green Version]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.C.; Prawira, A.; de Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef]
- Manzella, L.; Massimino, M.; Stella, S.; Tirro, E.; Pennisi, M.S.; Martorana, F.; Motta, G.; Vitale, S.R.; Puma, A.; Romano, C.; et al. Activation of the IGF Axis in Thyroid Cancer: Implications for Tumorigenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 3258. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Bible, K.C.; Chow, L.Q.M.; Gilbert, J.; Grande, C.; Worden, F.; Haddad, R. Management of treatment-related toxicities in advanced medullary thyroid cancer. Cancer Treat. Rev. 2018, 66, 64–73. [Google Scholar] [CrossRef]
- Massimino, M.; Tirro, E.; Stella, S.; Pennisi, M.S.; Vitale, S.R.; Puma, A.; Romano, C.; Romeo, M.A.; Gregorio, S.D.; Romeo, M.A.; et al. Targeting BCL-2 as a Therapeutic Strategy for Primary (p210)BCR-ABL1-positive B-ALL Cells. In Vivo 2020, 34, 511–516. [Google Scholar] [CrossRef] [Green Version]
- Tirro, E.; Massimino, M.; Romano, C.; Pennisi, M.S.; Stella, S.; Vitale, S.R.; Fidilio, A.; Manzella, L.; Parrinello, N.L.; Stagno, F.; et al. Chk1 Inhibition Restores Inotuzumab Ozogamicin Citotoxicity in CD22-Positive Cells Expressing Mutant p53. Front. Oncol. 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Zanardi, E.; Bregni, G.; de Braud, F.; Di Cosimo, S. Better Together: Targeted Combination Therapies in Breast Cancer. Semin. Oncol. 2015, 42, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2017, 14, 57–66. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
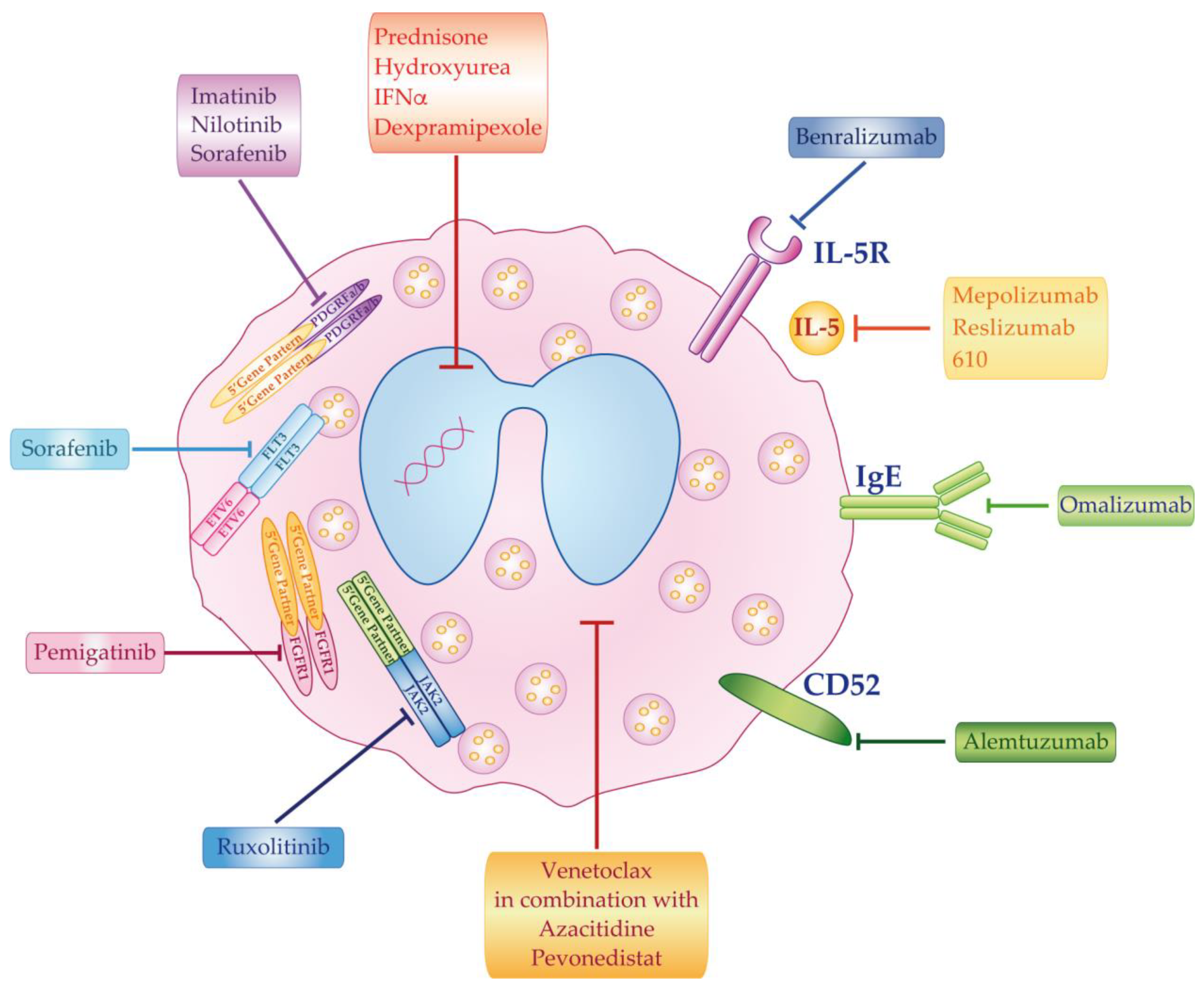{kind=link}
| Fusion Genes | |||
| Gene | Translocation | Gene | Translocation |
| PDGFRα | PDGFRβ | ||
| FIP1L1-PDGFRα | del(4)(q12;q12) | ETV6-PDGFRβ | t(5;12)(q33;p13) |
| BCR-PDGFRα | t(4;22)(q12;q11) | ||
| FGFR1 | JAK2 | ||
| ZMYM2-FGFR1 | t(8;13)(p11.2;q12.1) | PCM1-JAK2 | t(8;9)(p22;p24) |
| CNTRL-FGFR1 | t(8;9)(p11.2;q33.2) | BCR-JAK2 | t(9;22)(p24;q11.2) |
| BCR-FGFR1 | t(8;22)(p11.2;q11.2) | ETV6-JAK2 | t(9;12)(p24;p13) |
| Other Genes | |||
| ETV6-FLT3 | t(12;13)(p13;q12) | ||
| ETV6-ABL1 | t(9;12)(q34;p13) | ||
| Receptor Rearrangements | |||
| T Cell receptor rearrangement | |||
| Mutated Genes | |||
| Genes | Percentage of mutation | Genes | Percentage of mutation |
| RUNX1 | 83% | SETBP1 | 22% |
| ASXL1 | 43% | CBL | 14% |
| TET2 | 36% | NOTCH1 | 14% |
| EZH2 | 29% | ||
| Drug | Mechanism of Action | Dose | Target Neoplasm |
|---|---|---|---|
| Corticosteroids | |||
| Prednisone | Slow and prevent end-organ damage | 1 mg/kg daily | HES |
| Cytotoxic agents | |||
| Hydroxyurea | Inhibit DNA synthesis | 500–1000 mg/daily | HES (+ corticosteroids); Steroid non-responders. |
| IFNα | Inhibit cell growth and induct apoptosis | Initiation: 1 million units tiw * Escalation: 3–4 million units tiw * | HES (+ corticosteroids); HES & CEL, NOS refractory to other therapies; Lymphocyte-variant hypereosinophilia. |
| Targeted therapies | |||
| TKIs | |||
| Imatinib | Inhibit both TGFb and PDGF-R pathway | 100–400 mg/daily | PDGFRα rearranged; PDGRFβ rearranged; Alternate PDGRFβ fusions; Selected cases HES and CEL, NOS. |
| Ruxolitinib | Inhibit dysregulated JAK/STAT signalling pathway | 20 mg PO BID | Eosinophilic leukemia with the PCM1-JAK2 fusion [t(8;9)(p22;p24)]. |
| Sorafenib | Inhibit several kinases involved in both tumour cell proliferation and angiogenesis | 400 mg/twice daily | FIP1L1-PDGFRα rearranged pts with T674I mutation; FLT3-rearranged cases. |
| Monoclonal antibodies | |||
| Anti-IL-5 | |||
| Mepolizumab | Inhibit binding of IL-5 to the α chain of the IL-5R | 100–300 mg every 4 weeks | Eosinophilic asthma and eosinophilic granulomatosis with polyangiitis. |
| Reslizumab | Inhibit the proliferation of eosinophils by binding to the α chain of the IL-5R | 1 mg/kg | Eosinophilic asthma and eosinophilic esophagitis. |
| Anti-IL-5R | |||
| Benralizumab | Inhibit hetero-oligomerization of α and β subunits of IL-5R | 30 mg by subcutaneous injection every 4 weeks | Severe asthma. |
| Anti-IgE | |||
| Omalizumab | Inhibit release of cytokines such as IL-4, IL-5, and IL-13; block unbound IgE. | dose/frequency calculated bases on weight per serum IgE | Eosinophilic disorders, in particular asthma/nasal polyps |
| Anti-CD52 | |||
| Alemtuzumab | Mediate the lysis of CD52+ cells | 5–30 mg 1 to 3 times weekly | Refractory HES pts. |
| Drug | Combination | Target | Design | Patients | Identifier | Phase | Status |
|---|---|---|---|---|---|---|---|
| Corticosteroids | |||||||
| Prednisone | - | - | Single Group Assignment; Open Label | 100 | NCT01524536 | Phase IV | Recruiting |
| Dexpramipexole | - | - | Non-Randomized; Single Group Assignment; Open Label | 15 | NCT02101138 | Phase II | Unknown |
| Targeted therapies | |||||||
| TKIs | |||||||
| Imatinib | Ruxolitinib | FIP1L1-PDGFRα & PDGFRβ | Non-Randomized; Sequential Assignment; Open Label | 60 | NCT00044304 | Phase II | Recruiting |
| Nilotinib | - | FIP1L1-PDGFRα & PDGFRβ | NP | NP | NCT04498871 | NP | Available |
| Ruxolitinib | - | BCR-JAK2 Fusion Protein Expression | Single Group Assignment; Open Label | 25 | NCT03801434 | Phase II | Not yet recruiting |
| Monoclonal antibodies | |||||||
| Mepolizumab | IL-5 | NP | NP | NCT00244686 | NP | Available | |
| 610 * | Placebo | IL-5 | Randomized; Parallel Assignment | 52 | NCT04445038 | Phase I | Recruiting |
| Benralizumab | - | IL-5R | Multicentre; randomised; double-blind; parallel Assignment | 120 | NCT04191304 | Phase III | Not yet recruiting |
| Placebo | Randomized; Parallel Assignment | 22 | NCT02130882 | Phase II/III | Active, not recruiting | ||
| Chemotherapy | |||||||
| Venetoclax | Azacitidine, Pevonedistat | - | Single Group Assignment; Open Label | 40 | NCT03862157 | Phase I/II | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, S.; Massimino, M.; Manzella, L.; Pennisi, M.S.; Tirrò, E.; Romano, C.; Vitale, S.R.; Puma, A.; Tomarchio, C.; Gregorio, S.D.; et al. Molecular Pathogenesis and Treatment Perspectives for Hypereosinophilia and Hypereosinophilic Syndromes. Int. J. Mol. Sci. 2021, 22, 486. https://doi.org/10.3390/ijms22020486
Stella S, Massimino M, Manzella L, Pennisi MS, Tirrò E, Romano C, Vitale SR, Puma A, Tomarchio C, Gregorio SD, et al. Molecular Pathogenesis and Treatment Perspectives for Hypereosinophilia and Hypereosinophilic Syndromes. International Journal of Molecular Sciences. 2021; 22(2):486. https://doi.org/10.3390/ijms22020486
Chicago/Turabian StyleStella, Stefania, Michele Massimino, Livia Manzella, Maria Stella Pennisi, Elena Tirrò, Chiara Romano, Silvia Rita Vitale, Adriana Puma, Cristina Tomarchio, Sandra Di Gregorio, and et al. 2021. "Molecular Pathogenesis and Treatment Perspectives for Hypereosinophilia and Hypereosinophilic Syndromes" International Journal of Molecular Sciences 22, no. 2: 486. https://doi.org/10.3390/ijms22020486
APA StyleStella, S., Massimino, M., Manzella, L., Pennisi, M. S., Tirrò, E., Romano, C., Vitale, S. R., Puma, A., Tomarchio, C., Gregorio, S. D., Palumbo, G. A., & Vigneri, P. (2021). Molecular Pathogenesis and Treatment Perspectives for Hypereosinophilia and Hypereosinophilic Syndromes. International Journal of Molecular Sciences, 22(2), 486. https://doi.org/10.3390/ijms22020486