Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion

 , and
, and {kind=link}
{kind=link}
{kind=link}
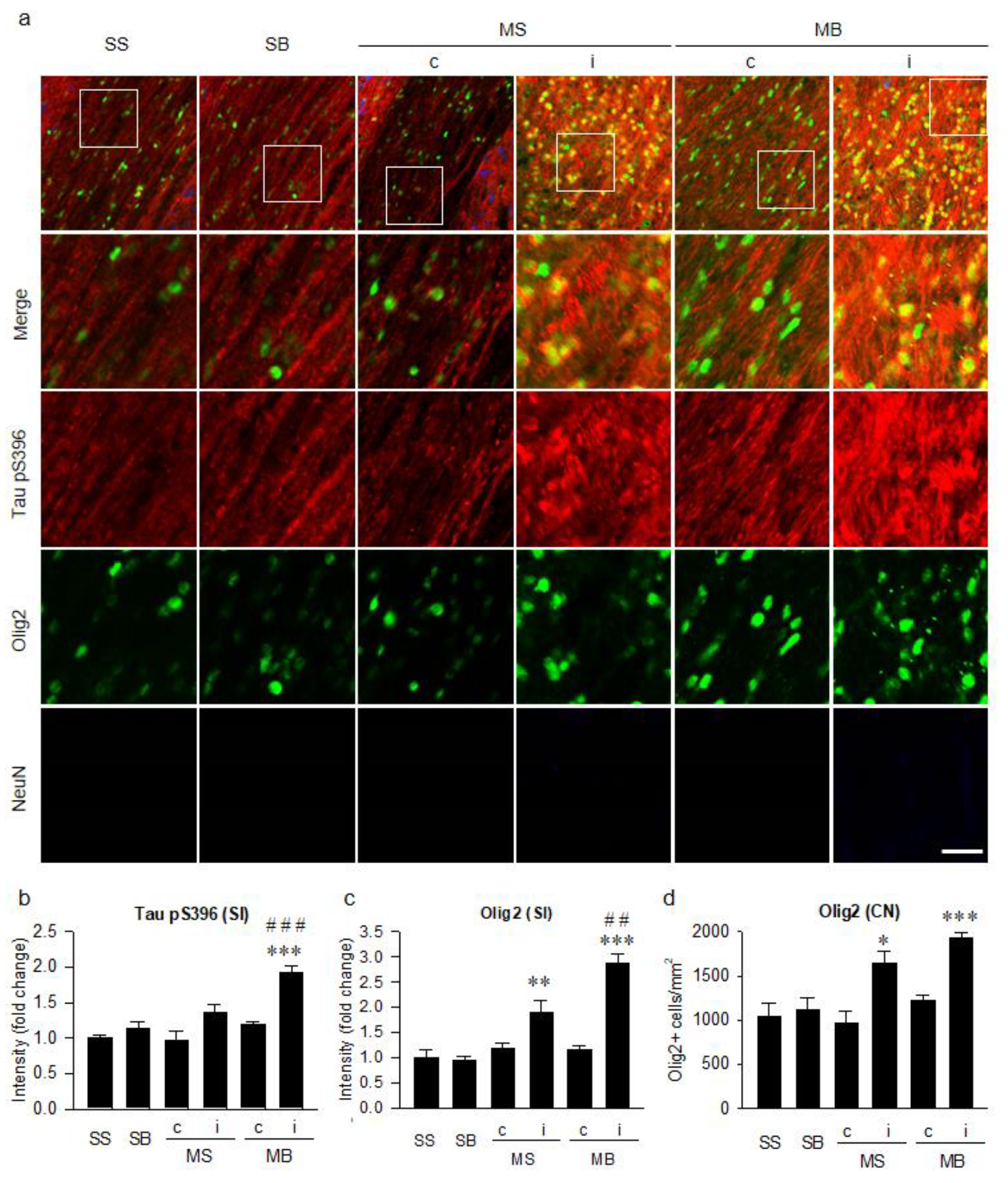{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Experimental Groups
2.2. Infarct Volume
2.3. Cognitive Impairments
2.4. Tauopathy in the Cortex
2.5. Tauopathy in the Corpus Callosum
2.6. Histological Correlation of Tauopathy
2.7. Parenchymal Infiltration of Cerebrospinal Fluid (CSF) Tracers
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
4.3. Neurological Function Test
4.4. Novel Object Tests
4.5. Tissue Preparation
4.6. Cresyl Violet Staining and Infarct Volume
4.7. Immunohistochemistry
4.8. Intracisternal Injection of CSF Tracers
4.9. Quantitative Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PSD | Post-stroke dementia |
| CCH | Chronic cerebral hypoperfusion |
| MCAO | Middle cerebral artery occlusion |
| BCCAo | Permanent occlusion of bilateral common carotid arteries |
| AQP4 | Aquaporin 4 |
| MB | MCAO + BCCAo |
| MS | MCAO + sham BCCAo |
| SB | Sham MCAO + BCCAo |
| SS | Sham MCAO + sham BCCAo |
| NOR | Novel object recognition test |
| NOL | Novel object location test |
| GFAP | Glial fibrillary acidic protein |
| MBP | Myelin basic protein |
| MAP2 | Microtubule associated protein 2 |
| CSF | Cerebrospinal fluid |
| DC | Dorsal cortex |
| VC | Ventral cortex |
| CG | Cingulate gyrus |
| BF | Basal forebrain |
| ISF | Interstitial fluid |
| FITC | Fluorescein isothiocyanate |
| ROI | Region of interest |
| PBS | Phosphate-buffered saline |
| PFA | Paraformaldehyde |
| SI | Signal intensity |
| CN | Cell number |
References
- Venketasubramanian, N.; Yoon, B.W.; Pandian, J.; Navarro, J.C. Stroke Epidemiology in South, East, and South-East Asia: A Review. J. Stroke 2017, 19, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L. Have Stroke Neurologists Entered the Arena of Stroke-Related Cognitive Dysfunctions? Not Yet, but They Should! Stroke 2017, 48, 1441–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, V.C.; Lam, B.Y.; Wong, A.; Ko, H.; Markus, H.S.; Wong, L.K. Early-onset and delayed-onset poststroke dementia—Revisiting the mechanisms. Nat. Rev. Neurol. 2017, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.R.; Kim, D.H.; Back, D.B.; Kang, C.H.; Moon, W.J.; Han, J.S.; Choi, D.H.; Kwon, K.J.; Shin, C.Y.; Kim, B.R.; et al. Characterization of White Matter Injury in a Rat Model of Chronic Cerebral Hypoperfusion. Stroke 2016, 47, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.R.; Kwon, K.J.; Park, S.H.; Jeon, W.K.; Han, S.H.; Kim, H.Y.; Han, J.S. Alternations of Septal-hippocampal System in the Adult Wistar Rat with Spatial Memory Impairments Induced by Chronic Cerebral Hypoperfusion. Exp. Neurobiol. 2011, 20, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.R.; Lee, S.R.; Han, J.S.; Woo, S.K.; Kim, K.M.; Choi, D.H.; Kwon, K.J.; Han, S.H.; Shin, C.Y.; Lee, J.; et al. Synergistic memory impairment through the interaction of chronic cerebral hypoperfusion and amlyloid toxicity in a rat model. Stroke 2011, 42, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Back, D.B.; Kwon, K.J.; Choi, D.H.; Shin, C.Y.; Lee, J.; Han, S.H.; Kim, H.Y. Chronic cerebral hypoperfusion induces post-stroke dementia following acute ischemic stroke in rats. J. Neuroinflamm. 2017, 14, 216. [Google Scholar] [CrossRef]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.; Dickson, D.W. Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 27–48. [Google Scholar] [CrossRef]
- Zhao, Y.; Gu, J.H.; Dai, C.L.; Liu, Q.; Iqbal, K.; Liu, F.; Gong, C.X. Chronic cerebral hypoperfusion causes decrease of O-GlcNAcylation, hyperphosphorylation of tau and behavioral deficits in mice. Front. Aging Neurosci. 2014, 6, 10. [Google Scholar] [CrossRef]
- Qiu, L.; Ng, G.; Tan, E.K.; Liao, P.; Kandiah, N.; Zeng, L. Chronic cerebral hypoperfusion enhances Tau hyperphosphorylation and reduces autophagy in Alzheimer’s disease mice. Sci. Rep. 2016, 6, 23964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliff, J.J.; Nedergaard, M. Is there a cerebral lymphatic system? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, E.N.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Wang, M.; Zeppenfeld, D.M.; Venkataraman, A.; Plog, B.A.; Liao, Y.; Deane, R.; Nedergaard, M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J. Neurosci. 2013, 33, 18190–18199. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Gao, T.; Luo, Y.; Chen, X.; Gao, G.; Gao, X.; Zhou, Y.; Dai, J. Transient focal cerebral ischemia/reperfusion induces early and chronic axonal changes in rats: Its importance for the risk of Alzheimer’s disease. PLoS ONE 2012, 7, e33722. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Yang, S.; Liu, R.; Simpkins, J.W. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004, 1022, 30–38. [Google Scholar] [CrossRef]
- Pluta, R.; Ulamek-Koziol, M.; Januszewski, S.; Czuczwar, S.J. Tau Protein Dysfunction after Brain Ischemia. J. Alzheimers Dis. 2018, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Dewar, D.; Underhill, S.M.; Goldberg, M.P. Oligodendrocytes and ischemic brain injury. J. Cereb. Blood Flow. Metab. 2003, 23, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, N.; Maki, T.; Pham, L.D.; Hayakawa, K.; Seo, J.H.; Mandeville, E.T.; Mandeville, J.B.; Kim, K.W.; Lo, E.H.; Arai, K. Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke 2013, 44, 3516–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buee, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, R.J.; Perry, G. Tau Biology, Tauopathy, Traumatic Brain Injury, and Diagnostic Challenges. J. Alzheimers Dis. 2019, 67, 447–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, E.A.; Yatsushiro, K.; McCulloch, J.; Dewar, D. Rapid alteration of tau in oligodendrocytes after focal ischemic injury in the rat: Involvement of free radicals. J. Cereb. Blood Flow. Metab. 1997, 17, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Irving, E.A.; Bentley, D.L.; Parsons, A.A. Assessment of white matter injury following prolonged focal cerebral ischaemia in the rat. Acta Neuropathol. 2001, 102, 627–635. [Google Scholar] [CrossRef]
- Buffo, A.; Vosko, M.R.; Erturk, D.; Hamann, G.F.; Jucker, M.; Rowitch, D.; Gotz, M. Expression pattern of the transcription factor Olig2 in response to brain injuries: Implications for neuronal repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18183–18188. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Nogawa, S.; Suzuki, S.; Dembo, T.; Kosakai, A. Upregulation of oligodendrocyte progenitor cells associated with restoration of mature oligodendrocytes and myelination in peri-infarct area in the rat brain. Brain Res. 2003, 989, 172–179. [Google Scholar] [CrossRef]
- McQueen, J.; Reimer, M.M.; Holland, P.R.; Manso, Y.; McLaughlin, M.; Fowler, J.H.; Horsburgh, K. Restoration of oligodendrocyte pools in a mouse model of chronic cerebral hypoperfusion. PLoS ONE 2014, 9, e87227. [Google Scholar] [CrossRef] [Green Version]
- Bonfanti, E.; Gelosa, P.; Fumagalli, M.; Dimou, L.; Vigano, F.; Tremoli, E.; Cimino, M.; Sironi, L.; Abbracchio, M.P. The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis. 2017, 8, e2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkat, P.; Chopp, M.; Zacharek, A.; Cui, C.; Zhang, L.; Li, Q.; Lu, M.; Zhang, T.; Liu, A.; Chen, J. White matter damage and glymphatic dysfunction in a model of vascular dementia in rats with no prior vascular pathologies. Neurobiol. Aging 2017, 50, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeppenfeld, D.M.; Simon, M.; Haswell, J.D.; D’Abreo, D.; Murchison, C.; Quinn, J.F.; Grafe, M.R.; Woltjer, R.L.; Kaye, J.; Iliff, J.J. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017, 74, 91–99. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Enger, R.; Hansson, H.A.; Nagelhus, E.A.; Eide, P.K. Loss of perivascular aquaporin-4 in idiopathic normal pressure hydrocephalus. Glia 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Patel, T.K.; Hochgrafe, K.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Mandelkow, E.M.; Holtzman, D.M. Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol. Neurodegener. 2015, 10, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, J.; Wang, L.; Lu, M.; Chopp, M. Treatment of stroke in rat with intracarotid administration of marrow stromal cells. Neurology 2001, 56, 1666–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Back, D.B.; Choi, B.-R.; Han, J.-S.; Kwon, K.J.; Choi, D.-H.; Shin, C.Y.; Lee, J.; Kim, H.Y. Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion. Int. J. Mol. Sci. 2020, 21, 6929. https://doi.org/10.3390/ijms21186929
Back DB, Choi B-R, Han J-S, Kwon KJ, Choi D-H, Shin CY, Lee J, Kim HY. Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion. International Journal of Molecular Sciences. 2020; 21(18):6929. https://doi.org/10.3390/ijms21186929
Chicago/Turabian StyleBack, Dong Bin, Bo-Ryoung Choi, Jung-Soo Han, Kyoung Ja Kwon, Dong-Hee Choi, Chan Young Shin, Jongmin Lee, and Hahn Young Kim. 2020. "Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion" International Journal of Molecular Sciences 21, no. 18: 6929. https://doi.org/10.3390/ijms21186929
APA StyleBack, D. B., Choi, B. -R., Han, J. -S., Kwon, K. J., Choi, D. -H., Shin, C. Y., Lee, J., & Kim, H. Y. (2020). Characterization of Tauopathy in a Rat Model of Post-Stroke Dementia Combining Acute Infarct and Chronic Cerebral Hypoperfusion. International Journal of Molecular Sciences, 21(18), 6929. https://doi.org/10.3390/ijms21186929