RET Receptor Tyrosine Kinase: Role in Neurodegeneration, Obesity, and Cancer



{kind=link}
{kind=link}
Abstract
:1. Introduction
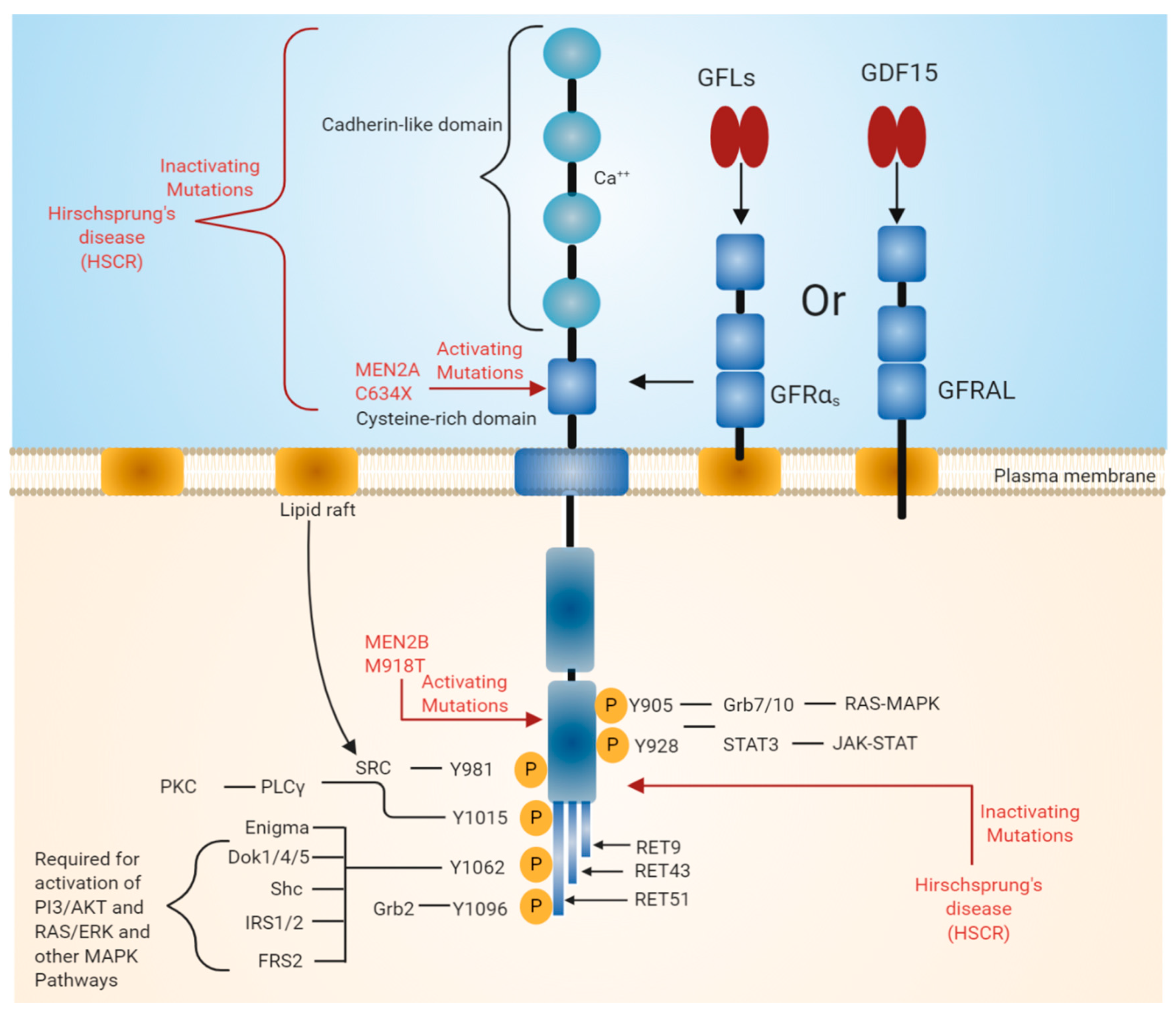
2. RET Receptor Tyrosine Kinase
3. Role of RET in Various Disease States
3.1. Normal Function of RET in DA Neurons and Its Implication in Parkinson’s Disease
3.2. Retinitis Pigmentosa and Other Eye Diseases
3.3. Hirschsprung Disease (HSCR)
3.4. Neuropathic Pain
3.5. Role of RET in the Non-Homeostatic Regulation of Body Weight
3.6. Role of RET in Cancer
3.6.1. Oncogenic Potential of Constitutively Active Oncogenic Forms of RET
3.6.2. Oncogenic Potential of Wild-Type RET
4. Targeting RET with Small Molecule for the Treatment of Diseases
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAv | Adeno-associated virus |
| ARTN | Artemin |
| BBB | Blood–brain barrier |
| CERE-120 | Adeno-associated virus-encoded neurturin |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinases |
| EsR | Estrogen receptor |
| GFLs | GDNF family ligands |
| GDNF | Glial cell line-derived neurotrophic factors |
| GFRAL | GDNF family receptor alpha-like |
| GFRα | GDNF family receptor alpha |
| GDF15 | Growth differentiation factor-15 |
| HSCR | Hirschsprung |
| MAPK | Mitogen-activated protein kinases |
| MEN | Multiple endocrine neoplasia |
| NRTN | Neurturin |
| NP | Neuropathic pain |
| PSPN | Persephin |
| PTC | Papillary thyroid carcinoma |
| PD | Parkinson’s disease |
| RETRP | Rearranged in transfectionRetinitis Pigmentosa |
| SNpc | Substantia nigra pars compacta |
References
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Menacho, I.; Barnouin, K.; Barry, R.; Borg, A.; Orme, M.; Chauhan, R.; Mouilleron, S.; Martínez-Torres, R.J.; Meier, P.; McDonald, N.Q. RET Functions as a Dual-Specificity Kinase that Requires Allosteric Inputs from Juxtamembrane Elements. Cell Rep. 2016, 17, 3319–3332. [Google Scholar] [CrossRef] [Green Version]
- Jmaeff, S.; Sidorova, Y.; Nedev, H.; Saarma, M.; Saragovi, H.U. Small-molecule agonists of the RET receptor tyrosine kinase activate biased trophic signals that are influenced by the presence of GFRa1 co-receptors. J. Biol. Chem. 2020, jbc.RA119.011802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esseghir, S.; Todd, S.K.; Hunt, T.; Poulsom, R.; Plaza-Menacho, I.; Reis-Filho, J.S.; Isacke, C.M. A Role for Glial Cell–Derived Neurotrophic Factor–Induced Expression by Inflammatory Cytokines and RET/GFRα1 Receptor Up-regulation in Breast Cancer. Cancer Res. 2007, 67, 11732–11741. [Google Scholar] [CrossRef] [Green Version]
- Knowles, P.P.; Murray-Rust, J.; Kjær, S.; Scott, R.P.; Hanrahan, S.; Santoro, M.; Ibáñez, C.F.; McDonald, N.Q. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Ritz, J.; Cooper, G.M. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985, 42, 581–588. [Google Scholar] [CrossRef]
- Takahashi, M.; Cooper, G.M. Ret transforming gene encodes a fusion protein homologous to tyrosine kinases. Mol. Cell. Biol. 1987, 7, 1378–1385. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Asai, N.; Iwashita, T.; Isomura, T.; Miyazaki, K.; Matsuyama, M. Characterization of the ret proto-oncogene products expressed in mouse L cells. Oncogene 1993, 8, 2925–2929. [Google Scholar]
- Runeberg-Roos, P.; Saarma, P.M. Neurotrophic factor receptor RET: Structure, cell biology, and inherited diseases. Ann. Med. 2007, 39, 572–580. [Google Scholar] [CrossRef]
- Anders, J.; Kjar, S.; Ibáñez, C.F. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. J. Biol. Chem. 2001, 276, 35808–35817. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, C.; Asai, N.; Murakami, H.; Iwashita, T.; Iwata, Y.; Horibe, K.; Klein, R.D.; Rosenthal, A.; Takahashi, M. Calcium-dependent Ret activation by GDNF and neurturin. Oncogene 1998, 16, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Weering, D.H.J.; Moen, T.C.; Braakman, I.; Baas, P.D.; Bos, J.L. Expression of the Receptor Tyrosine Kinase Ret on the Plasma Membrane Is Dependent on Calcium. J. Biol. Chem. 1998, 273, 12077–12081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahira, T.; Ishizaka, Y.; Itoh, F.; Sugimura, T.; Nagao, M. Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene 1990, 5, 97–102. [Google Scholar]
- Carter, M.T.; Yome, J.L.; Marcil, M.N.; Martin, C.A.; Vanhorne, J.B.; Mulligan, L.M. Conservation of RET proto-oncogene splicing variants and implications for RET isoform function. CGR 2001, 95, 169–176. [Google Scholar] [CrossRef]
- de Graaff, E.; Srinivas, S.; Kilkenny, C.; D’Agati, V.; Mankoo, B.S.; Costantini, F.; Pachnis, V. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 2001, 15, 2433–2444. [Google Scholar] [CrossRef] [Green Version]
- Tsai, V.W.W.; Husaini, Y.; Sainsbury, A.; Brown, D.A.; Breit, S.N. The MIC-1/GDF15-GFRAL Pathway in Energy Homeostasis: Implications for Obesity, Cachexia, and Other Associated Diseases. Cell Metab. 2018, 28, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Unsicker, K.; Spittau, B.; Krieglstein, K. The multiple facets of the TGF-β family cytokine growth/differentiation factor-15/macrophage inhibitory cytokine-1. Cytokine Growth Factor Rev. 2013, 24, 373–384. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Manié, S.; Santoro, M.; Fusco, A.; Billaud, M. The RET receptor: Function in development and dysfunction in congenital malformation. Trends Genet. 2001, 17, 580–589. [Google Scholar] [CrossRef]
- Bespalov, M.M.; Saarma, M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol. Sci. 2007, 28, 68–74. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Mätlik, K.; Paveliev, M.; Lindahl, M.; Piranen, E.; Milbrandt, J.; Arumäe, U.; Saarma, M.; Bespalov, M.M. Persephin signaling through GFRalpha1: The potential for the treatment of Parkinson’s disease. Mol. Cell. Neurosci. 2010, 44, 223–232. [Google Scholar] [CrossRef]
- Baloh, R.H.; Tansey, M.G.; Lampe, P.A.; Fahrner, T.J.; Enomoto, H.; Simburger, K.S.; Leitner, M.L.; Araki, T.; Johnson, E.M.; Milbrandt, J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron 1998, 21, 1291–1302. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chang, C.-C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjær, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.-N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Crawley, S.; Chen, M.; Ayupova, D.A.; Lindhout, D.A.; Higbee, J.; Kutach, A.; Joo, W.; Gao, Z.; Fu, D.; et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 2017, 550, 255–259. [Google Scholar] [CrossRef]
- Pierchala, B.A.; Milbrandt, J.; Johnson, E.M., Jr. Glial Cell Line-Derived Neurotrophic Factor-Dependent Recruitment of Ret into Lipid Rafts Enhances Signaling by Partitioning Ret from Proteasome-Dependent Degradation. J. Neurosci. 2006, 26, 2777. [Google Scholar] [CrossRef] [Green Version]
- Tsui, C.C.; Gabreski, N.A.; Hein, S.J.; Pierchala, B.A. Lipid Rafts Are Physiologic Membrane Microdomains Necessary for the Morphogenic and Developmental Functions of Glial Cell Line-Derived Neurotrophic Factor In Vivo. J. Neurosci. 2015, 35, 13233. [Google Scholar] [CrossRef] [Green Version]
- Murakumo, Y.; Jijiwa, M.; Asai, N.; Ichihara, M.; Takahashi, M. RET and neuroendocrine tumors. Pituitary 2006, 9, 179–192. [Google Scholar] [CrossRef]
- Santoro, M.; Melillo, R.M.; Carlomagno, F.; Vecchio, G.; Fusco, A. Minireview: RET: Normal and Abnormal Functions. Endocrinology 2004, 145, 5448–5451. [Google Scholar] [CrossRef]
- Avantaggiato, V.; Dathan, N.A.; Grieco, M.; Fabien, N.; Lazzaro, D.; Fusco, A.; Simeone, A.; Santoro, M. Developmental expression of the RET protooncogene. Cell Growth Differ. 1994, 5, 305. [Google Scholar] [PubMed]
- Pachnis, V.; Mankoo, B.; Costantini, F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development 1993, 119, 1005–1017. [Google Scholar] [PubMed]
- Attié-Bitach, T.; Abitbol, M.; Gérard, M.; Delezoide, A.-L.; Augé, J.; Pelet, A.; Amiel, J.; Pachnis, V.; Munnich, A.; Lyonnet, S.; et al. Expression of the RET proto-oncogene in human Embryos. Am. J. Med Genet. 1998, 80, 481–486. [Google Scholar] [CrossRef]
- Kramer, E.R.; Liss, B. GDNF–Ret signaling in midbrain dopaminergic neurons and its implication for Parkinson disease. FEBS Lett. 2015, 589, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Mijatovic, J.; Airavaara, M.; Planken, A.; Auvinen, P.; Raasmaja, A.; Piepponen, T.P.; Costantini, F.; Ahtee, L.; Saarma, M. Constitutive Ret Activity in Knock-In Multiple Endocrine Neoplasia Type B Mice Induces Profound Elevation of Brain Dopamine Concentration via Enhanced Synthesis and Increases the Number of TH-Positive Cells in the Substantia Nigra. J. Neurosci. 2007, 27, 4799–4809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Liu, F.; Miao, C.; Li, Q.; Zhang, Z.; Xiao, P.; Su, L.; Yu, K.; Chen, X.; Zhang, F.; et al. RET somatic mutations are underrecognized in Hirschsprung disease. Genet. Med. 2018, 20, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Durbec, P.; Marcos-Gutierrez, C.V.; Kilkenny, C.; Grigoriou, M.; Wartiowaara, K.; Suvanto, P.; Smith, D.; Ponder, B.; Costantini, F.; Saarma, M.; et al. GDNF signalling through the Ret receptor tyrosine kinase. Nature 1996, 381, 789–793. [Google Scholar] [CrossRef]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar] [CrossRef]
- Marcos, C.; Pachnis, V. The effect of the ret- mutation on the normal development of the central and parasympathetic nervous systems. Int. J. Dev. Biol. 1996, 40 (Suppl. 1), 137S–138S. [Google Scholar]
- Jain, S.; Golden, J.P.; Wozniak, D.; Pehek, E.; Johnson, E.M.; Milbrandt, J. RET Is Dispensable for Maintenance of Midbrain Dopaminergic Neurons in Adult Mice. J. Neurosci. 2006, 26, 11230–11238. [Google Scholar] [CrossRef] [Green Version]
- Kramer, E.R.; Aron, L.; Ramakers, G.M.J.; Seitz, S.; Zhuang, X.; Beyer, K.; Smidt, M.P.; Klein, R. Absence of Ret Signaling in Mice Causes Progressive and Late Degeneration of the Nigrostriatal System. PLoS Biol. 2007, 5, e39. [Google Scholar] [CrossRef] [Green Version]
- Mijatovic, J.; Patrikainen, O.; Yavich, L.; Airavaara, M.; Ahtee, L.; Saarma, M.; Piepponen, T.P. Characterization of the striatal dopaminergic neurotransmission in MEN2B mice with elevated cerebral tissue dopamine. J. Neurochem. 2008, 105, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Mahato, A.K.; Kopra, J.; Renko, J.-M.; Visnapuu, T.; Korhonen, I.; Pulkkinen, N.; Bespalov, M.M.; Domanskyi, A.; Ronken, E.; Piepponen, T.P.; et al. Glial cell line-derived neurotrophic factor receptor Rearranged during transfection agonist supports dopamine neurons in Vitro and enhances dopamine release In Vivo. Mov. Disord. 2020, 35, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Mahato, A.K.; Renko, J.-M.; Kopra, J.; Visnapuu, T.; Korhonen, I.; Pulkkinen, N.; Bespalov, M.; Ronken, E.; Piepponen, T.P.; Voutilainen, M.; et al. GDNF receptor agonist supports dopamine neurons in vitro and protects their function in animal model of Parkinson’s. Neuroscience 2019. [Google Scholar]
- Chmielarz, P.; Er, Ş.; Konovalova, J.; Bandrés, L.; Hlushchuk, I.; Albert, K.; Panhelainen, A.; Luk, K.; Airavaara, M.; Domanskyi, A. GDNF/RET signaling pathway activation eliminates Lewy Body pathology in midbrain dopamine neurons. bioRxiv 2019, 752899. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Horger, B.A.; Nishimura, M.C.; Armanini, M.P.; Wang, L.-C.; Poulsen, K.T.; Rosenblad, C.; Kirik, D.; Moffat, B.; Simmons, L.; Johnson, E.; et al. Neurturin Exerts Potent Actions on Survival and Function of Midbrain Dopaminergic Neurons. J. Neurosci. 1998, 18, 4929–4937. [Google Scholar] [CrossRef] [Green Version]
- Zihlmann, K.B.; Ducray, A.D.; Schaller, B.; Huber, A.W.; Krebs, S.H.; Andres, R.H.; Seiler, R.W.; Meyer, M.; Widmer, H.R. The GDNF family members neurturin, artemin and persephin promote the morphological differentiation of cultured ventral mesencephalic dopaminergic neurons. Brain Res. Bull. 2005, 68, 42–53. [Google Scholar] [CrossRef]
- Hoffer, B.J.; Hoffman, A.; Bowenkamp, K.; Huettl, P.; Hudson, J.; Martin, D.; Lin, L.F.; Gerhardt, G.A. Glial cell line-derived neurotrophic factor reverses toxin-induced injury to midbrain dopaminergic neurons in vivo. Neurosci. Lett. 1994, 182, 107–111. [Google Scholar] [CrossRef]
- Bowenkamp, K.E.; Hoffman, A.F.; Gerhardt, G.A.; Henry, M.A.; Biddle, P.T.; Hoffer, B.J.; Granholm, A.-C.E. Glial cell line-derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J. Comp. Neurol. 1995, 355, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.D.; Valverde, J.; Alexi, T.; Poulsen, K.; Moffat, B.; Vandlen, R.A.; Rosenthal, A.; Hefti, F. Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature 1995, 373, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Gash, D.M.; Zhang, Z.; Ovadia, A.; Cass, W.A.; Yi, A.; Simmerman, L.; Russell, D.; Martin, D.; Lapchak, P.A.; Collins, F.; et al. Functional recovery in parkinsonian monkeys treated with GDNF. Nature 1996, 380, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Rosenblad, C.; Bjorklund, A.; Mandel, R.J. Long-term rAAV-mediated gene transfer of GDNF in the rat Parkinson’s model: Intrastriatal but not intranigral transduction promotes functional regeneration in the lesioned nigrostriatal system. J. Neurosci. 2000, 20, 4686–4700. [Google Scholar] [CrossRef]
- Kordower, J.H.; Emborg, M.E.; Bloch, J.; Ma, S.Y.; Chu, Y.; Leventhal, L.; McBride, J.; Chen, E.Y.; Palfi, S.; Roitberg, B.Z.; et al. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 2000, 290, 767–773. [Google Scholar] [CrossRef]
- Oiwa, Y.; Yoshimura, R.; Nakai, K.; Itakura, T. Dopaminergic neuroprotection and regeneration by neurturin assessed by using behavioral, biochemical and histochemical measurements in a model of progressive Parkinson’s disease. Brain Res. 2002, 947, 271–283. [Google Scholar] [CrossRef]
- Runeberg-Roos, P.; Piccinini, E.; Penttinen, A.-M.; Mätlik, K.; Heikkinen, H.; Kuure, S.; Bespalov, M.M.; Peränen, J.; Garea-Rodríguez, E.; Fuchs, E.; et al. Developing therapeutically more efficient Neurturin variants for treatment of Parkinson’s disease. Neurobiol. Dis. 2016, 96, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef]
- Slevin, J.T.; Gerhardt, G.A.; Smith, C.D.; Gash, D.M.; Kryscio, R.; Young, B. Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J. Neurosurg. 2005, 102, 216–222. [Google Scholar] [CrossRef]
- Marks, W.J.; Ostrem, J.L.; Verhagen, L.; Starr, P.A.; Larson, P.S.; Bakay, R.A.; Taylor, R.; Cahn-Weiner, D.A.; Stoessl, A.J.; Olanow, C.W.; et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol. 2008, 7, 400–408. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Whone, A.L.; Boca, M.; Luz, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Extended Treatment with Glial Cell Line-Derived Neurotrophic Factor in Parkinson’s Disease. J. Parkinsons’s Dis. 2019, 9, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef] [Green Version]
- Bartus, R.T.; Johnson, E.M. Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 1: Where have we been and what have we learned? Neurobiol. Dis. 2017, 97, 156–168. [Google Scholar] [CrossRef]
- Mahato, A.K.; Sidorova, Y.A. Glial cell line-derived neurotrophic factors (GFLs) and small molecules targeting RET receptor for the treatment of pain and Parkinson’s disease. Cell Tissue Res. 2020. [Google Scholar] [CrossRef]
- Datta, R.; Waheed, A.; Bonapace, G.; Shah, G.N.; Sly, W.S. Pathogenesis of retinitis pigmentosa associated with apoptosis-inducing mutations in carbonic anhydrase IV. Proc. Natl. Acad. Sci. USA 2009, 106, 3437–3442. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.G.; Wang, X.; Chavis, P.; Kuriyan, A.E.; Abariga, S.A. Vitamin A and fish oils for preventing the progression of retinitis pigmentosa. Cochrane Database Syst. Rev. 2020, 6, CD008428. [Google Scholar] [CrossRef]
- Muhammad Imran Qadir, S. Retinitis Pigmentosa: Pathophysiology and its Management. Fortune J. Health Sci. 2018, 1, 19–25. [Google Scholar] [CrossRef]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis Pigmentosa: Genes and Disease Mechanisms. Curr. Genomics 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Dalkara, D.; Kolstad, K.D.; Guerin, K.I.; Hoffmann, N.V.; Visel, M.; Klimczak, R.R.; Schaffer, D.V.; Flannery, J.G. AAV Mediated GDNF Secretion From Retinal Glia Slows Down Retinal Degeneration in a Rat Model of Retinitis Pigmentosa. Mol. Ther. 2011, 19, 1602–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee Sanftner, L.H.; Abel, H.; Hauswirth, W.W.; Flannery, J.G. Glial cell line derived neurotrophic factor delays photoreceptor degeneration in a transgenic rat model of retinitis pigmentosa. Mol. Ther. 2001, 4, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Allocca, M.; Vicino, U.D.; Petrillo, M.; Carlomagno, F.; Domenici, L.; Auricchio, A. Constitutive and AP20187-Induced Ret Activation in Photoreceptors Does Not Protect from Light-Induced Damage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5199–5206. [Google Scholar] [CrossRef] [PubMed]
- Jmaeff, S.; Sidorova, Y.; Lippiatt, H.; Barcelona, P.F.; Nedev, H.; Saragovi, L.M.; Hancock, M.A.; Saarma, M.; Saragovi, H.U. Small-Molecule Ligands that Bind the RET Receptor Activate Neuroprotective Signals Independent of but Modulated by Coreceptor GFRα1. Mol. Pharmacol. 2020, 98, 1–12. [Google Scholar] [CrossRef]
- Flachsbarth, K.; Jankowiak, W.; Kruszewski, K.; Helbing, S.; Bartsch, S.; Bartsch, U. Pronounced synergistic neuroprotective effect of GDNF and CNTF on axotomized retinal ganglion cells in the adult mouse. Exp. Eye Res. 2018, 176, 258–265. [Google Scholar] [CrossRef]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.J.; Le, Y.-Z. Müller Glia Are a Major Cellular Source of Survival Signals for Retinal Neurons in Diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Zhang, H.; Zhu, M.; Le, Y.-Z. Critical Role of Trophic Factors in Protecting Müller Glia: Implications to Neuroprotection in Age-Related Macular Degeneration, Diabetic Retinopathy, and Anti-VEGF Therapies. Adv. Exp. Med. Biol. 2019, 1185, 469–473. [Google Scholar] [CrossRef]
- Wu, L.; Xiao, P.; Li, Q.; Zhang, Z.; Wang, H.; Jiang, Q.; Li, L. Altered expression of AKT1 and P38A in the colons of patients with Hirschsprung’s disease. Pediatric Surg. Int. 2020, 36, 719–725. [Google Scholar] [CrossRef]
- De Falco, V.; Carlomagno, F.; Li, H.; Santoro, M. The molecular basis for RET tyrosine-kinase inhibitors in thyroid cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 307–318. [Google Scholar] [CrossRef]
- Tomuschat, C.; Puri, P. RET gene is a major risk factor for Hirschsprung’s disease: A meta-analysis. Pediatric Surg. Int. 2015, 31, 701–710. [Google Scholar] [CrossRef]
- Porokuokka, L.L.; Virtanen, H.T.; Lindén, J.; Sidorova, Y.; Danilova, T.; Lindahl, M.; Saarma, M.; Andressoo, J.-O. Gfra1 Underexpression Causes Hirschsprung’s Disease and Associated Enterocolitis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 655–678. [Google Scholar] [CrossRef] [Green Version]
- Lui, V.C.H.; Samy, E.T.; Sham, M.H.; Mulligan, L.M.; Tam, P.K.H. Glial Cell Line–Derived Neurotrophic Factor Family Receptors Are Abnormally Expressed in Aganglionic Bowel of a Subpopulation of Patients with Hirschsprung’s Disease. Lab. Investig. 2002, 82, 703–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IASP. Available online: https://www.iasp-pain.org/Advocacy/GYAP.aspx?ItemNumber=5054 (accessed on 12 August 2020).
- Yawn, B.P.; Wollan, P.C.; Weingarten, T.N.; Watson, J.C.; Hooten, W.M.; Melton, L.J. The prevalence of neuropathic pain: Clinical evaluation compared with screening tools in a community population. Pain Med. 2009, 10, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, F.; van de Walle, J.; Zurowska, A.; Gimpel, C.; van Hoeck, K.; Drozdz, D.; Montini, G.; Bagdasorova, I.V.; Sorof, J.; Sugg, J.; et al. Efficacy, safety and pharmacokinetics of candesartan cilexetil in hypertensive children from 1 to less than 6 years of age. J. Hypertens. 2010, 28, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Ossipov, M.H. Growth factors and neuropathic pain. Curr. Pain Headache Rep. 2011, 15, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, K.J.; Viisanen, H.; Ahlström, F.H.G.; Jokinen, V.; Sidorova, Y.A.; Suleymanova, I.; Rauhala, P.V.; Kalso, E.A.; Lilius, T.O. Morphine-3-glucuronide causes antinociceptive cross-tolerance to morphine and increases spinal substance P expression. Eur. J. Pharmacol. 2020, 875, 173021. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpaa, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: Systematic review, meta-analysis and updated NeuPSIG recommendations. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.L.; Boucher, T.J.; Armanini, M.P.; Poulsen, K.T.; Michael, G.J.; Priestley, J.V.; Phillips, H.S.; McMahon, S.B.; Shelton, D.L. The glial cell line-derived neurotrophic factor family receptor components are differentially regulated within sensory neurons after nerve injury. J. Neurosci. 2000, 20, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Josephson, A.; Widenfalk, J.; Trifunovski, A.; Widmer, H.R.; Olson, L.; Spenger, C. GDNF and NGF family members and receptors in human fetal and adult spinal cord and dorsal root ganglia. J. Comp. Neurol. 2001, 440, 204–217. [Google Scholar] [CrossRef]
- Gardell, L.R.; Wang, R.; Ehrenfels, C.; Ossipov, M.H.; Rossomando, A.J.; Miller, S.; Buckley, C.; Cai, A.K.; Tse, A.; Foley, S.F.; et al. Multiple actions of systemic artemin in experimental neuropathy. Nat. Med. 2003, 9, 1383–1389. [Google Scholar] [CrossRef]
- Wang, R.; Rossomando, A.; Sah, D.W.Y.; Ossipov, M.H.; King, T.; Porreca, F. Artemin induced functional recovery and reinnervation after partial nerve injury. Pain 2014, 155, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, T.J.; Okuse, K.; Bennett, D.L.; Munson, J.B.; Wood, J.N.; McMahon, S.B. Potent analgesic effects of GDNF in neuropathic pain states. Science 2000, 290, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Backonja, M.; Williams, L.; Miao, X.; Katz, N.; Chen, C. Safety and efficacy of neublastin in painful lumbosacral radiculopathy: A randomized, double-blinded, placebo-controlled phase 2 trial using Bayesian adaptive design (the SPRINT trial). Pain 2017, 158, 1802–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolan, P.E.; O’Neill, G.; Versage, E.; Rana, J.; Tang, Y.; Galluppi, G.; Aycardi, E. First-In-Human, Double-Blind, Placebo-Controlled, Randomized, Dose-Escalation Study of BG00010, a Glial Cell Line-Derived Neurotrophic Factor Family Member, in Subjects with Unilateral Sciatica. PLoS ONE 2015, 10, e0125034. [Google Scholar] [CrossRef]
- Lippoldt, E.K.; Elmes, R.R.; McCoy, D.D.; Knowlton, W.M.; McKemy, D.D. Artemin, a glial cell line-derived neurotrophic factor family member, induces TRPM8-dependent cold pain. J. Neurosci. 2013, 33, 12543–12552. [Google Scholar] [CrossRef]
- Lippoldt, E.K.; Ongun, S.; Kusaka, G.K.; McKemy, D.D. Inflammatory and neuropathic cold allodynia are selectively mediated by the neurotrophic factor receptor GFRα3. Proc. Natl. Acad. Sci. USA 2016, 113, 4506–4511. [Google Scholar] [CrossRef] [Green Version]
- Usoskin, D.; Furlan, A.; Islam, S.; Abdo, H.; Lönnerberg, P.; Lou, D.; Hjerling-Leffler, J.; Haeggström, J.; Kharchenko, O.; Kharchenko, P.V.; et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015, 18, 145–153. [Google Scholar] [CrossRef]
- Clemmensen, C.; Müller, T.D.; Woods, S.C.; Berthoud, H.-R.; Seeley, R.J.; Tschöp, M.H. Gut-Brain Cross-Talk in Metabolic Control. Cell 2017, 168, 758–774. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Huen, S.C.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.-D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525.e12. [Google Scholar] [CrossRef] [Green Version]
- Quartu, M.; Serra, M.P.; Boi, M.; Ferretti, M.T.; Lai, M.L.; Del Fiacco, M. Tissue distribution of Ret, GFRalpha-1, GFRalpha-2 and GFRalpha-3 receptors in the human brainstem at fetal, neonatal and adult age. Brain Res. 2007, 1173, 36–52. [Google Scholar] [CrossRef]
- Mulligan, L.M. GDNF and the RET Receptor in Cancer: New Insights and Therapeutic Potential. Front. Physiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Carlomagno, F. Central Role of RET in Thyroid Cancer. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, C. Multiple Endocrine Neoplasia Type 2. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Runeberg-Roos, P.; Virtanen, H.; Saarma, M. RET(MEN 2B) is active in the endoplasmic reticulum before reaching the cell surface. Oncogene 2007, 26, 7909–7915. [Google Scholar] [CrossRef] [PubMed]
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers (Basel) 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Sangwan, V.; Park, M. Receptor tyrosine kinases: Role in cancer progression. Curr. Oncol. 2006, 13, 191–193. [Google Scholar]
- Nikiforov, Y.E. RET/PTC rearrangement in thyroid tumors. Endocr. Pathol. 2002, 13, 3–16. [Google Scholar] [CrossRef]
- Thomas, G.A.; Bunnell, H.; Cook, H.A.; Williams, E.D.; Nerovnya, A.; Cherstvoy, E.D.; Tronko, N.D.; Bogdanova, T.I.; Chiappetta, G.; Viglietto, G.; et al. High Prevalence of RET/PTC Rearrangements in Ukrainian and Belarussian Post-Chernobyl Thyroid Papillary Carcinomas: A Strong Correlation between RET/PTC3 and the Solid-Follicular Variant. J. Clin. Endocrinol. Metab. 1999, 84, 4232–4238. [Google Scholar] [CrossRef]
- Kohno, T.; Ichikawa, H.; Totoki, Y.; Yasuda, K.; Hiramoto, M.; Nammo, T.; Sakamoto, H.; Tsuta, K.; Furuta, K.; Shimada, Y.; et al. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 2012, 18, 375–377. [Google Scholar] [CrossRef]
- Lipson, D.; Capelletti, M.; Yelensky, R.; Otto, G.; Parker, A.; Jarosz, M.; Curran, J.A.; Balasubramanian, S.; Bloom, T.; Brennan, K.W.; et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med. 2012, 18, 382–384. [Google Scholar] [CrossRef]
- Ju, Y.S.; Lee, W.-C.; Shin, J.-Y.; Lee, S.; Bleazard, T.; Won, J.-K.; Kim, Y.T.; Kim, J.-I.; Kang, J.-H.; Seo, J.-S. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012, 22, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Meka, D.P.; Müller-Rischart, A.K.; Nidadavolu, P.; Mohammadi, B.; Motori, E.; Ponna, S.K.; Aboutalebi, H.; Bassal, M.; Annamneedi, A.; Finckh, B.; et al. Parkin cooperates with GDNF/RET signaling to prevent dopaminergic neuron degeneration. J. Clin. Investig. 2015, 125, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Brown, P.H. The rearranged during transfection/papillary thyroid carcinoma tyrosine kinase is an estrogen-dependent gene required for the growth of estrogen receptor positive breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 487–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, L.; Pietilä, E.; Öhman, T.; Liu, X.; Mahato, A.K.; Sidorova, Y.; Lehti, K.; Saarma, M.; Varjosalo, M. PTPRA Phosphatase Regulates GDNF-Dependent RET Signaling and Inhibits the RET Mutant MEN2A Oncogenic Potential. iScience 2020, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche-Guenther, R.; Witzel, F.; Sieber, A.; Herr, R.; Schmidt, N.; Braun, S.; Brummer, T.; Sers, C.; Blüthgen, N. Strong negative feedback from Erk to Raf confers robustness to MAPK signalling. Mol. Syst. Biol. 2011, 7, 489. [Google Scholar] [CrossRef]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [Green Version]
- Arkun, Y.; Yasemi, M. Dynamics and control of the ERK signaling pathway: Sensitivity, bistability, and oscillations. PLoS ONE 2018, 13, e0195513. [Google Scholar] [CrossRef]
- Tansey, M.G.; Baloh, R.H.; Milbrandt, J.; Johnson, E.M. GFRα-Mediated Localization of RET to Lipid Rafts Is Required for Effective Downstream Signaling, Differentiation, and Neuronal Survival. Neuron 2000, 25, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Gil, Z.; Cavel, O.; Kelly, K.; Brader, P.; Rein, A.; Gao, S.P.; Carlson, D.L.; Shah, J.P.; Fong, Y.; Wong, R.J. Paracrine Regulation of Pancreatic Cancer Cell Invasion by Peripheral Nerves. J. Natl. Cancer Inst. 2010, 102, 107–118. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Xu, Q.; Lv, S.; Li, J.; Ma, Q. Role of glial cell line-derived neurotrophic factor in perineural invasion of pancreatic cancer. Biochim. Biophys. Acta (BBA) Rev. Cancer 2012, 1826, 112–120. [Google Scholar] [CrossRef]
- Ban, K.; Feng, S.; Shao, L.; Ittmann, M. RET signaling in prostate cancer. Clin. Cancer Res. 2017, 23, 4885–4896. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Cheng, Y.; Zhu, Q.; Yu, Z.; Wu, X.; Huang, K.; Zhou, M.; Han, S.; Zhang, Q. The Relationship between Over-expression of Glial Cell-derived Neurotrophic Factor and Its RET Receptor with Progression and Prognosis of Human Pancreatic Cancer. J. Int. Med. Res. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza-Menacho, I.; Morandi, A.; Robertson, D.; Pancholi, S.; Drury, S.; Dowsett, M.; Martin, L.-A.; Isacke, C.M. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene 2010, 29, 4648–4657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, A.; Martin, L.-A.; Gao, Q.; Pancholi, S.; Mackay, A.; Robertson, D.; Zvelebil, M.; Dowsett, M.; Plaza-Menacho, I.; Isacke, C.M. GDNF–RET Signaling in ER-Positive Breast Cancers Is a Key Determinant of Response and Resistance to Aromatase Inhibitors. Cancer Res. 2013, 73, 3783–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattelli, A.; Nalvarte, I.; Boulay, A.; Roloff, T.C.; Schreiber, M.; Carragher, N.; Macleod, K.K.; Schlederer, M.; Lienhard, S.; Kenner, L.; et al. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol. Med. 2013, 5, 1335–1350. [Google Scholar] [CrossRef] [PubMed]
- White, U.A.; Stephens, J.M. The gp130 Receptor Cytokine Family: Regulators of Adipocyte Development and Function. Curr. Pharm. Des. 2011, 17, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Boulay, A.; Breuleux, M.; Stephan, C.; Fux, C.; Brisken, C.; Fiche, M.; Wartmann, M.; Stumm, M.; Lane, H.A.; Hynes, N.E. The Ret Receptor Tyrosine Kinase Pathway Functionally Interacts with the ERα Pathway in Breast Cancer. Cancer Res. 2008, 68, 3743–3751. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Figueroa, M.; Bonilla, S.; Gutiérrez, F.; Pascual, A.; López-Barneo, J. GDNF Is Predominantly Expressed in the PV+ Neostriatal Interneuronal Ensemble in Normal Mouse and after Injury of the Nigrostriatal Pathway. J. Neurosci. 2012, 32, 864–872. [Google Scholar] [CrossRef]
- Ibáñez, C.F.; Paratcha, G.; Ledda, F. RET-independent signaling by GDNF ligands and GFRα receptors. Cell Tissue Res. 2020. [Google Scholar] [CrossRef]
- Paratcha, G.; Ledda, F.; Ibáñez, C.F. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003, 113, 867–879. [Google Scholar] [CrossRef] [Green Version]
- Turconi, G.; Kopra, J.; Võikar, V.; Kulesskaya, N.; Vilenius, C.; Piepponen, T.P.; Andressoo, J.-O. Chronic 2-Fold Elevation of Endogenous GDNF Levels Is Safe and Enhances Motor and Dopaminergic Function in Aged Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 831–842. [Google Scholar] [CrossRef]
- Villanueva, M.T. GDF15 tells the brain to lose weight. Nat. Rev. Drug Discov. 2017, 16, 827. [Google Scholar] [CrossRef] [PubMed]
- Bespalov, M.M.; Sidorova, Y.A.; Tumova, S.; Ahonen-Bishopp, A.; Magalhães, A.C.; Kulesskiy, E.; Paveliev, M.; Rivera, C.; Rauvala, H.; Saarma, M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011, 192, 153–169. [Google Scholar] [CrossRef] [Green Version]
- Sidorova, Y.A.; Bespalov, M.M.; Wong, A.W.; Kambur, O.; Jokinen, V.; Lilius, T.O.; Suleymanova, I.; Karelson, G.; Rauhala, P.V.; Karelson, M.; et al. A Novel Small Molecule GDNF Receptor RET Agonist, BT13, Promotes Neurite Growth from Sensory Neurons in Vitro and Attenuates Experimental Neuropathy in the Rat. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Viisanen, H.; Nuotio, U.; Kambur, O.; Mahato, A.K.; Jokinen, V.; Lilius, T.; Li, W.; Santos, H.A.; Karelson, M.; Rauhala, P.; et al. Novel RET agonist for the treatment of experimental neuropathies. Mol. Pain 2020, 16, 1744806920950866. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Tammiku-Taul, J.; García-Sosa, A.T.; Sidorova, Y.; Saarma, M.; Karelson, M. Molecular Dynamics Simulations of the Interactions between Glial Cell Line-Derived Neurotrophic Factor Family Receptor GFRα1 and Small-Molecule Ligands. ACS Omega 2018, 3, 11407–11414. [Google Scholar] [CrossRef] [Green Version]
- Amirouchene-Angelozzi, N.; Swanton, C.; Bardelli, A. Tumor Evolution as a Therapeutic Target. Cancer Discov. 2017, 7, 805–817. [Google Scholar] [CrossRef] [Green Version]
- Trupp, M.; Arenas, E.; Fainzilber, M.; Nilsson, A.-S.; Sieber, B.-A.; Grigoriou, M.; Kilkenny, C.; Salazar-Grueso, E.; Pachnis, V.; Arumäe, U.; et al. Functional receptor for GDNF encoded by the c- ret proto-oncogene. Nature 1996, 381, 785–789. [Google Scholar] [CrossRef]
- Cintrón-Colón, A.F.; Almeida-Alves, G.; Boynton, A.M.; Spitsbergen, J.M. GDNF synthesis, signaling, and retrograde transport in motor neurons. Cell Tissue Res. 2020. [Google Scholar] [CrossRef]
- Ron, D.; Janak, P.H. GDNF and addiction. Rev. Neurosci. 2005, 16, 277–285. [Google Scholar] [CrossRef]
- Reeben, M.; Laurikainen, A.; Hiltunen, J.O.; Castren, E.; Saarma, M. The messenger RNAs for both glial cell line-derived neurotrophic factor receptors, c-ret and GDNFR alpha, are induced in the rat brain in response to kainate-induced excitation. Neuroscience 1998, 83, 151–159. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahato, A.K.; Sidorova, Y.A. RET Receptor Tyrosine Kinase: Role in Neurodegeneration, Obesity, and Cancer. Int. J. Mol. Sci. 2020, 21, 7108. https://doi.org/10.3390/ijms21197108
Mahato AK, Sidorova YA. RET Receptor Tyrosine Kinase: Role in Neurodegeneration, Obesity, and Cancer. International Journal of Molecular Sciences. 2020; 21(19):7108. https://doi.org/10.3390/ijms21197108
Chicago/Turabian StyleMahato, Arun Kumar, and Yulia A. Sidorova. 2020. "RET Receptor Tyrosine Kinase: Role in Neurodegeneration, Obesity, and Cancer" International Journal of Molecular Sciences 21, no. 19: 7108. https://doi.org/10.3390/ijms21197108
APA StyleMahato, A. K., & Sidorova, Y. A. (2020). RET Receptor Tyrosine Kinase: Role in Neurodegeneration, Obesity, and Cancer. International Journal of Molecular Sciences, 21(19), 7108. https://doi.org/10.3390/ijms21197108