Autophagy Dynamics and Modulation in a Rat Model of Renal Ischemia-Reperfusion Injury

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
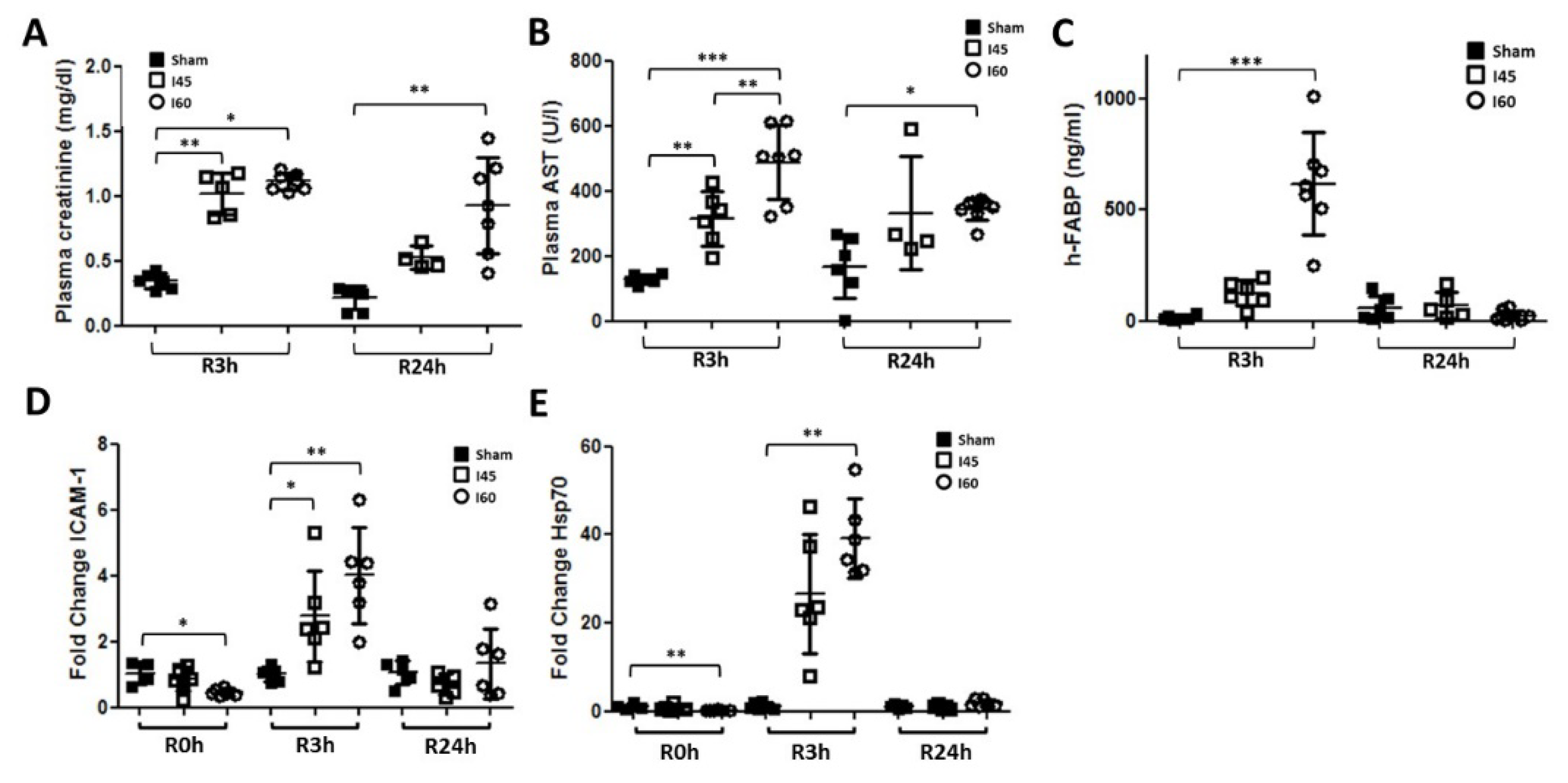
2.1. Transient Renal Injury Following Mild Ischemia
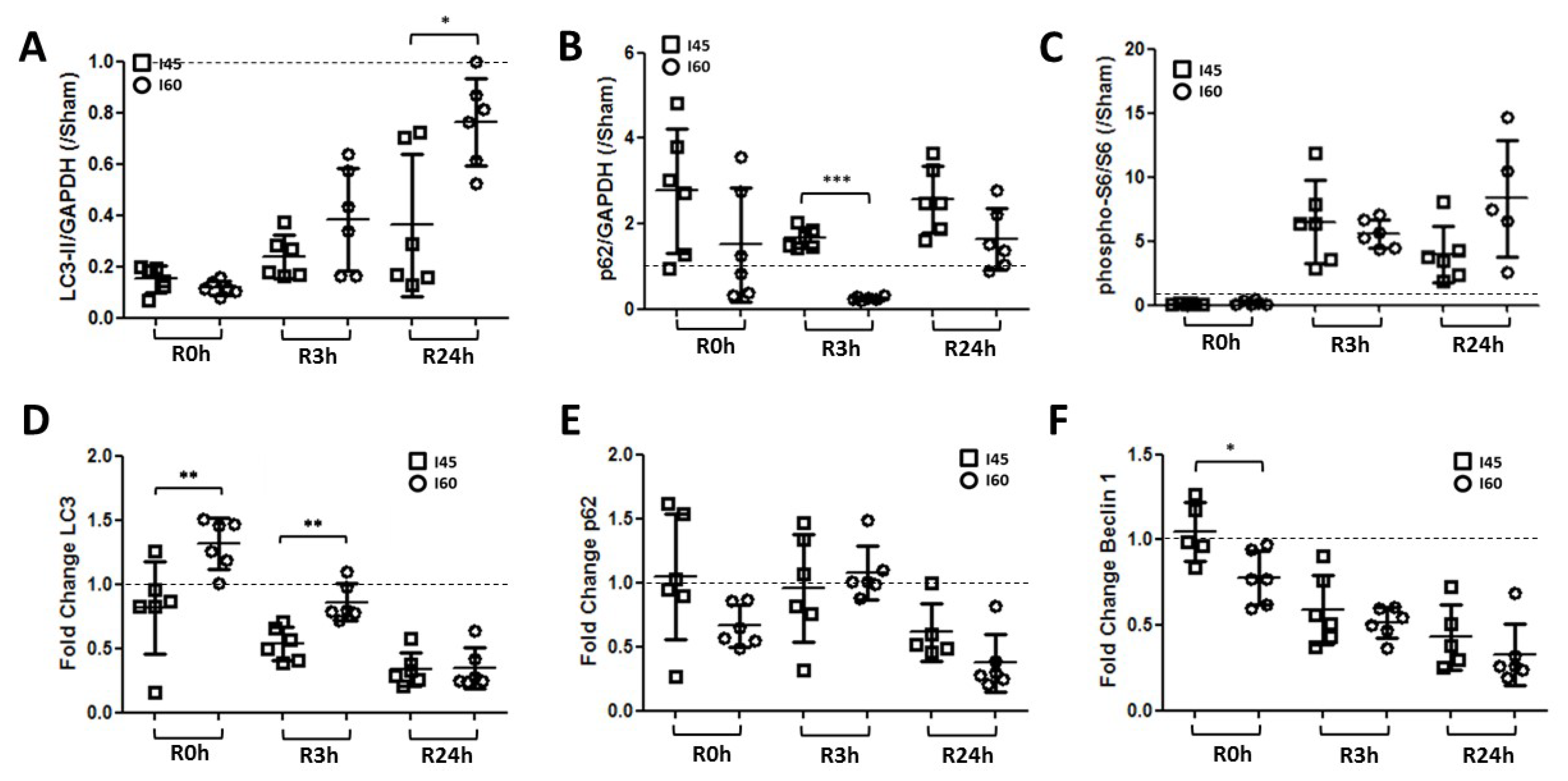
2.2. Autophagy Is Suppressed during Ischemia and Reperfusion Following Mild Ischemia
2.3. Apoptosis Is Enhanced during Reperfusion Following Mild Ischemia
2.4. More Severe Ischemia Increases Kidney Damage
2.5. More Autophagy upon Severe Compared to Mild Ischemia
2.6. Trehalose Stimulates Autophagy and Reduces IR Injury in the Kidney
3. Discussion
4. Materials and Methods
4.1. Ischemia-Reperfusion Injury Model
4.2. Experimental Groups
4.3. Kidney Function and Injury
4.4. Transmission Electron Microscopy
4.5. Western Blotting
4.6. Antibodies
4.7. Quantitative Real-Time Polymerase Chain Reaction
4.8. qPCR Taqman Probes
4.9. Terminal Deoxynucleotidyl Transferase End Labeling
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| AST | Aspartate transaminase |
| Bcl-2 | B-Cell lymphoma 2 |
| hFABP | heart-type fatty acid binding protein |
| Hsp70 | Heat shock protein 70 |
| ICAM | Intracellular adhesion molecule |
| IL | Interleukin |
| IR | Ischemia-reperfusion |
| LC3 | (microtubule-associated protein 1) Light chain 3 |
| mTOR | mammalian target of rapamycin |
| Sqstm1 | Sequestosome 1 |
| TEM | transmission electron microscopy |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| ULK-1 | Unc51-like kinase-1 |
References
- Bellomo, R.; Kellum, J.A.; Ronco, C. Acute kidney injury. Lancet 2012, 380, 756–766. [Google Scholar] [CrossRef]
- Lameire, N.; Bagga, A.; Cruz, D.; De Maeseneer, J.; Endre, Z.H.; Kellum, J.A.; Liu, K.D.; Mehta, R.L.; Pannu, N.; Van Biesen, W.; et al. Acute kidney injury: An increasing global concern. Lancet 2013, 382, 170–179. [Google Scholar] [CrossRef]
- Hoste, E.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerdá, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef] [PubMed]
- RENAL Replacement Therapy Study Investigators; Bellomo, R.; Cass, A.; Cole, L.; Finfer, S.; Gallagher, M.; Billot, L.; McArthur, C.; McGuinness, S.; Myburgh, J.A.; et al. Intensity of continuous renal-replacement therapy in critically ill patients. N. Engl. J. Med. 2009, 361, 1627–1638. [Google Scholar] [PubMed] [Green Version]
- Palevsky, P.M.; Zhang, J.H.; O’Connor, T.Z.; Chertow, G.M.; Crowley, S.T.; Choudhury, D.; Finkel, K.; Kellum, J.A.; Paganini, E.; Schein, R.M.H.; et al. Intensity of renal support in critically ill patients with acute kidney injury. N. Engl. J. Med. 2008, 359, 7–20. [Google Scholar] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Chawla, L.S.; Kimmel, P.L. Acute kidney injury and chronic kidney disease: An integrated clinical syndrome. Kidney Int. 2012, 82, 516–524. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Pallet, N.; Livingston, M.; Dong, Z. Emerging Functions of Autophagy in Kidney Transplantation. Am. J. Transplant. 2013, 14, 13–20. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daemen, M.A.; Veer, C.v.; Denecker, G.; Heemskerk, V.H.; Wolfs, T.G.A.M.; Clauss, M.; Vandenabeele, P.; Buurman, W.A. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J. Clin. Investig. 1999, 104, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daemen, M.A.; de Vries, B.; Buurman, W.A. Apoptosis and inflammation in renal reperfusion injury. Transplantation 2002, 73, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Hosgood, S.A.; Nicholson, M.L. Naked small interfering RNA of caspase-3 in preservation solution and autologous blood perfusate protects isolated ischemic porcine kidneys. Transplantation 2011, 91, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Nishihara, K.; Inui, K.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol. 2012, 696, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.T.; Shyue, S.K.; Lai, M.K. Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 2007, 84, 1183–1190. [Google Scholar] [CrossRef]
- Isaka, Y.; Suzuki, C.; Abe, T.; Okumi, M.; Ichimaru, N.; Imamura, R.; Kakuta, Y.; Matsui, I.; Takabatake, Y.; Rakugi, H.; et al. Bcl-2 protects tubular epithelial cells from ischemia/reperfusion injury by dual mechanisms. Transplant. Proc. 2009, 41, 52–54. [Google Scholar] [CrossRef]
- Yeh, C.H.; Hsu, S.P.; Yang, C.C.; Chien, C.T.; Wang, N.P. Hypoxic preconditioning reinforces HIF-alpha-dependent HSP70 signaling to reduce ischemic renal failure-induced renal tubular apoptosis and autophagy. Life Sci. 2010, 86, 115–123. [Google Scholar] [CrossRef]
- Wu, H.H.; Hsiao, T.Y.; Chien, C.T.; Lai, M.K. Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J. Biomed. Sci. 2009, 16, 19. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decuypere, J.P.; Pirenne, J.; Jochmans, I. Autophagy in Renal Ischemia-Reperfusion Injury: Friend or Foe? Am. J. Transplant. 2014, 14, 1464–1465. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Ceulemans, L.J.; Agostinis, P.; Monbaliu, D.; Naesens, M.; Pirenne, J.; Jochmans, I. Autophagy and the kidney: Implications for ischemia-reperfusion injury and therapy. Am. J. Kidney Dis. 2015, 66, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnaclliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 82, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Ceulemans, L.J.; Wylin, T.; Martinet, W.; Monbaliu, D.; Pirenne, J.; Jochmans, I. Plasmatic Villin 1 Is a Novel In Vivo Marker of Proximal Tubular Cell Injury During Renal Ischemia-Reperfusion. Transplantation 2017, 101, e330–e336. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, Y.; Goodenough, S.; Inoue, Y.; Dodd, P.R.; Wilce, P.A.; Matsumoto, I. Increased TUNEL positive cells in human alcoholic brains. Neurosci. Lett. 2003, 349, 201–205. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Biever, A.; Valjent, E.; Puighermanal, E. Ribosomal Protein S6 Phosphorylation in the Nervous System: From Regulation to Function. Front. Mol. Neurosci. 2015, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Klionsky, D.J. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. 2011, 13, 119–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bore, P.J.; Papatheofanis, I.; Sells, R.A. Adenosine triphosphate regeneration and function in the rat kidney following warm ischaemia. Transplantation 1979, 27, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Liu, K.; Luo, J.; Dong, Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am. J. Pathol. 2010, 176, 1181–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyagi, T.; Kusakari, Y.; Xiao, C.-Y.; Inouye, B.T.; Takahashi, M.; Scherrer-Crosbie, M.; Rosenzweig, A.; Hara, K.; Matsui, T. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am. J. Heart Physiol. Circ. Physiol. 2012, 303, H75–H85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Mulay, S.R.; Thomasova, D.; Ryu, M.; Anders, H.J. MDM2 (murine double minute 2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 2012, 81, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated Cell Death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701. [Google Scholar] [CrossRef]
- Belavgeni, A.; Meyer, C.; Stumpf, J.; Hugo, C.; Linkermann, A. Ferroptosis and Necroptosis in the Kidney. Cell Chem. Biol. 2020, 27, 446–462. [Google Scholar] [CrossRef]
- Van Erp, A.C.; Hoeksma, D.; Rebolledo, R.A.; Ottens, P.J.; Jochmans, I.; Monbaliu, D.; Pirenne, J.; Leuvenink, H.; Decuypere, J.-P. The Crosstalk between ROS and Autophagy in the Field of Transplantation Medicine. Oxidative Med. Cell. Longev. 2017, 2017, 7120962. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Pissas, G.; Antoniadi, G.; Liakopoulos, V.; Stefanidis, I. Cell Death Patterns Due to Warm Ischemia or Reperfusion in Renal Tubular Epithelial Cells Originating from Human, Mouse, or the Native Hibernator Hamster. Biology 2018, 7, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciarretta, S.; Yee, D.; Nagarajan, N.; Bianchi, F.; Saito, T.; Valenti, V.; Tong, M.; Del Re, M.P.; Vecchione, C.; Schirone, L.; et al. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 71, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Bando, T.; Yamada, T.; Sato, M.; Menjyu, T.; Aoyama, A.; Sato, T.; Chen, F.; Sonobe, M.; Omasa, M.; et al. Clinical application of ET-Kyoto solution for lung transplantation. Surg. Today 2015, 45, 439–443. [Google Scholar] [CrossRef]
- Zhao, X.; Koshiba, T.; Nakamura, T.; Tsuruyama, T.; Li, Y.; Bando, T.; Wada, H.; Tanaka, K. ET-Kyoto solution plus dibutyryl cyclic adenosine monophosphate is superior to University of Wisconsin solution in rat liver preservation. Cell Transplant. 2008, 17, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Jochmans, I.; Lerut, E.; Van Pelt, J.; Monbaliu, D.; Pirenne, J. Circulating AST, H-FABP, and NGAL are early and accurate biomarkers of graft injury and dysfunction in a preclinical model of kidney transplantation. Ann. Surg. 2011, 254, 784–791. [Google Scholar] [CrossRef]
- Shirakabe, A.; Kobayashi, N.; Hata, N.; Shinada, T.; Tomita, K.; Tsurumi, M.; Okazaki, H.; Matsushita, M.; Yamamoto, Y.; Yokoyama, S.; et al. The serum heart-type fatty acid-binding protein (HFABP) levels can be used to detect the presence of acute kidney injury on admission in patients admitted to the non-surgical intensive care unit. BMC Cardiovasc. Disord. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinet, W.; Timmermans, J.P.; De Meyer, G.R.Y. Methods to assess autophagy in situ—Transmission electron microscopy versus immunohistochemistry. Methods Enzymol. 2014, 543, 89–114. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Decuypere, J.-P.; Hutchinson, S.; Monbaliu, D.; Martinet, W.; Pirenne, J.; Jochmans, I. Autophagy Dynamics and Modulation in a Rat Model of Renal Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2020, 21, 7185. https://doi.org/10.3390/ijms21197185
Decuypere J-P, Hutchinson S, Monbaliu D, Martinet W, Pirenne J, Jochmans I. Autophagy Dynamics and Modulation in a Rat Model of Renal Ischemia-Reperfusion Injury. International Journal of Molecular Sciences. 2020; 21(19):7185. https://doi.org/10.3390/ijms21197185
Chicago/Turabian StyleDecuypere, Jean-Paul, Shawn Hutchinson, Diethard Monbaliu, Wim Martinet, Jacques Pirenne, and Ina Jochmans. 2020. "Autophagy Dynamics and Modulation in a Rat Model of Renal Ischemia-Reperfusion Injury" International Journal of Molecular Sciences 21, no. 19: 7185. https://doi.org/10.3390/ijms21197185
APA StyleDecuypere, J. -P., Hutchinson, S., Monbaliu, D., Martinet, W., Pirenne, J., & Jochmans, I. (2020). Autophagy Dynamics and Modulation in a Rat Model of Renal Ischemia-Reperfusion Injury. International Journal of Molecular Sciences, 21(19), 7185. https://doi.org/10.3390/ijms21197185