SPOTONE: Hot Spots on Protein Complexes with Extremely Randomized Trees via Sequence-Only Features



Abstract
:1. Introduction
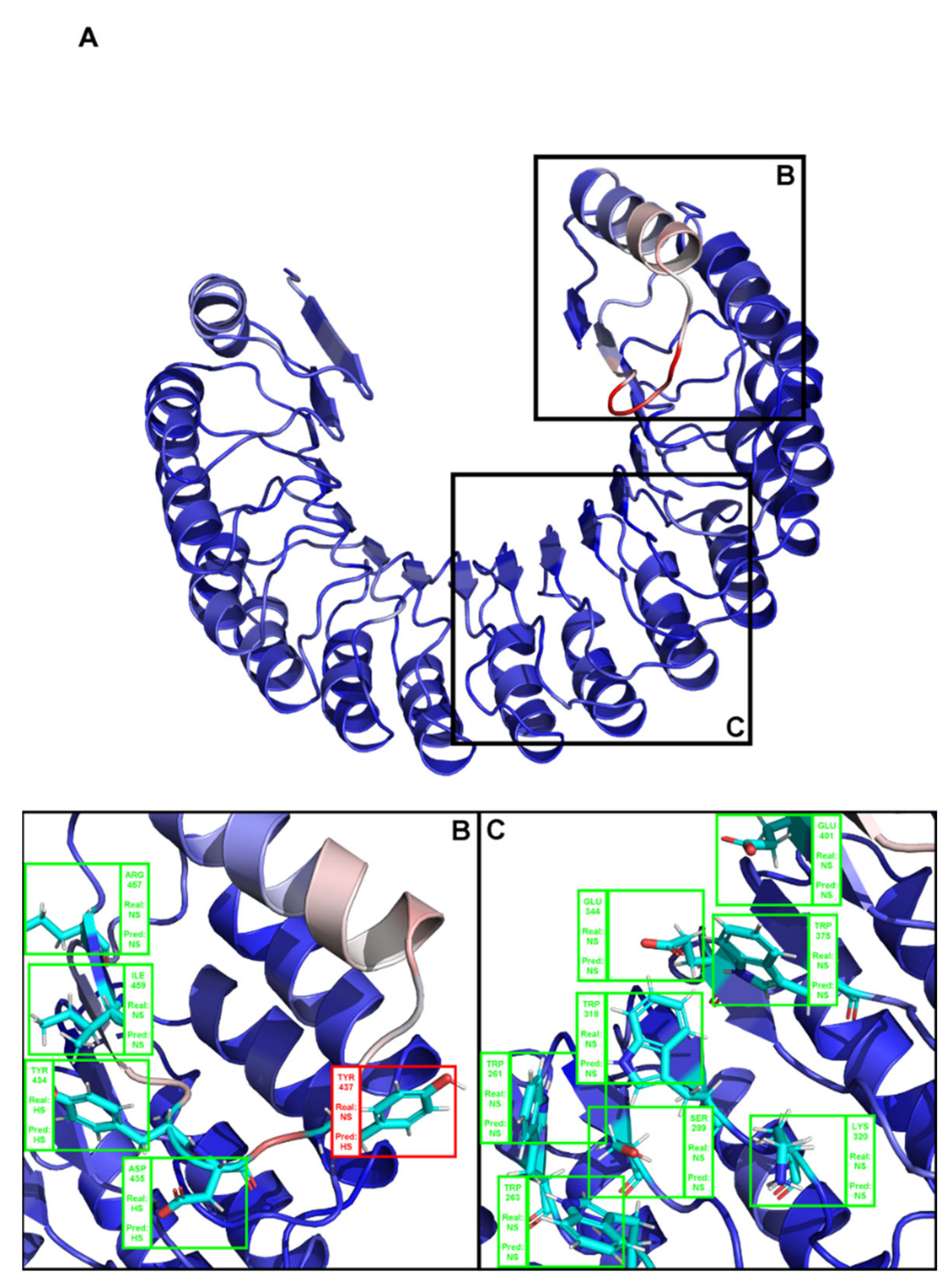
2. Results
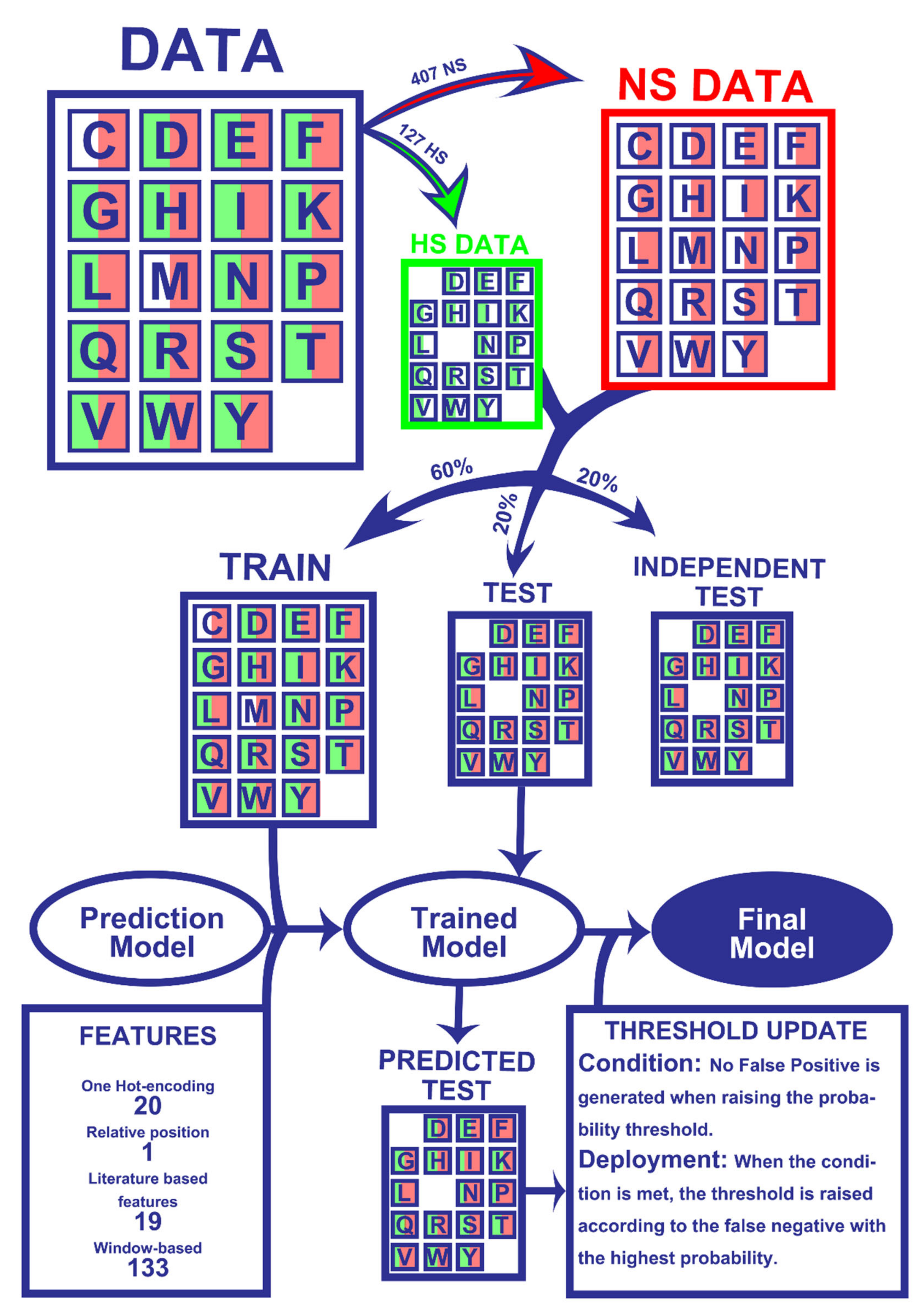
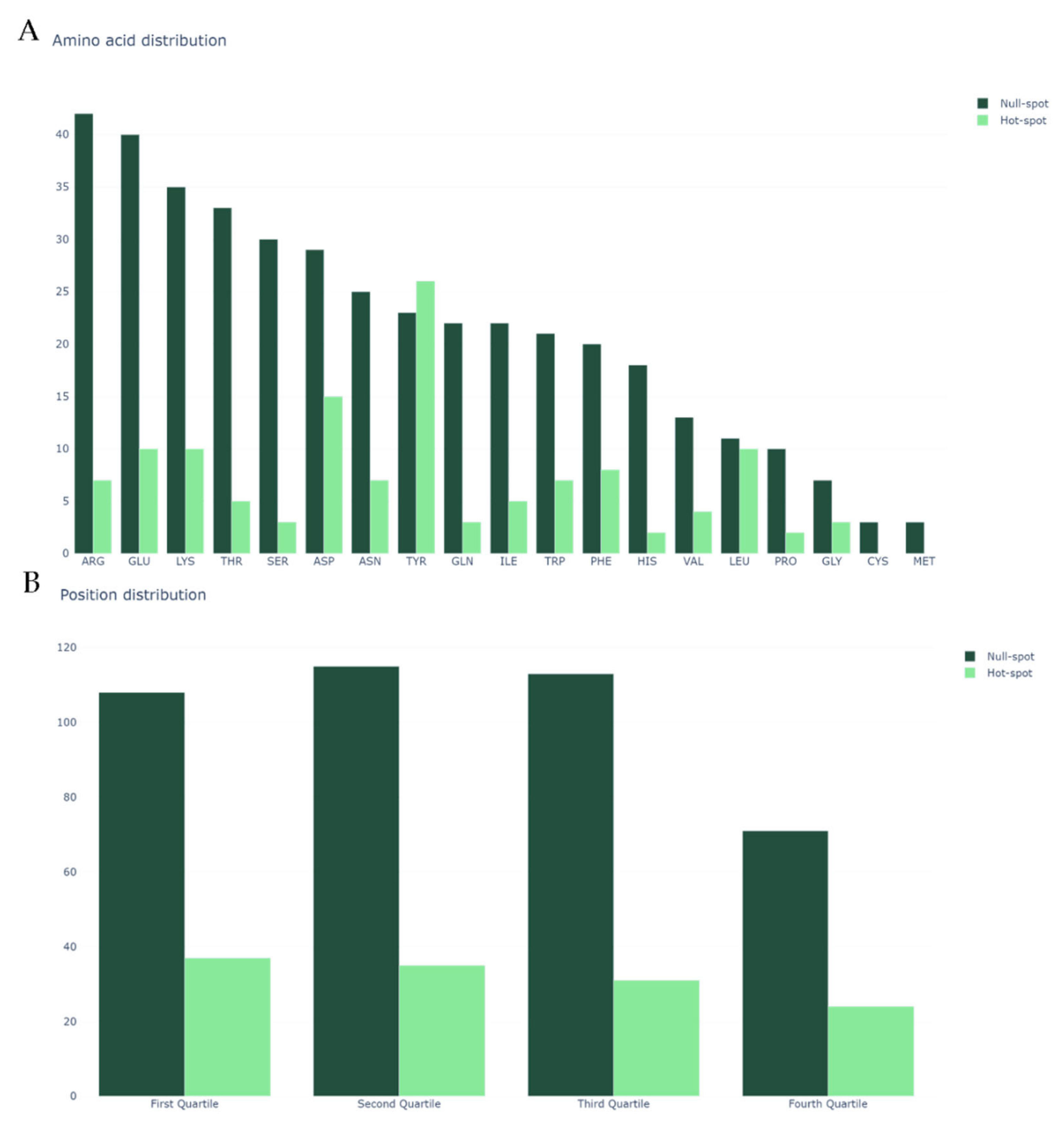
2.1. Dataset
2.2. Machine-Learning Algorithms
3. Discussion
4. Materials and Methods
4.1. Features
4.2. One-hot Encoding (20 Features)
4.3. Relative Position Feature (1 Feature)
4.4. Literature-Based Features (19 Features)
4.5. Window-Based Features (133 Features)
4.6. Machine-Learning Models Deployment
4.7. Model Evaluation
4.8. Prediction Probability Tuning
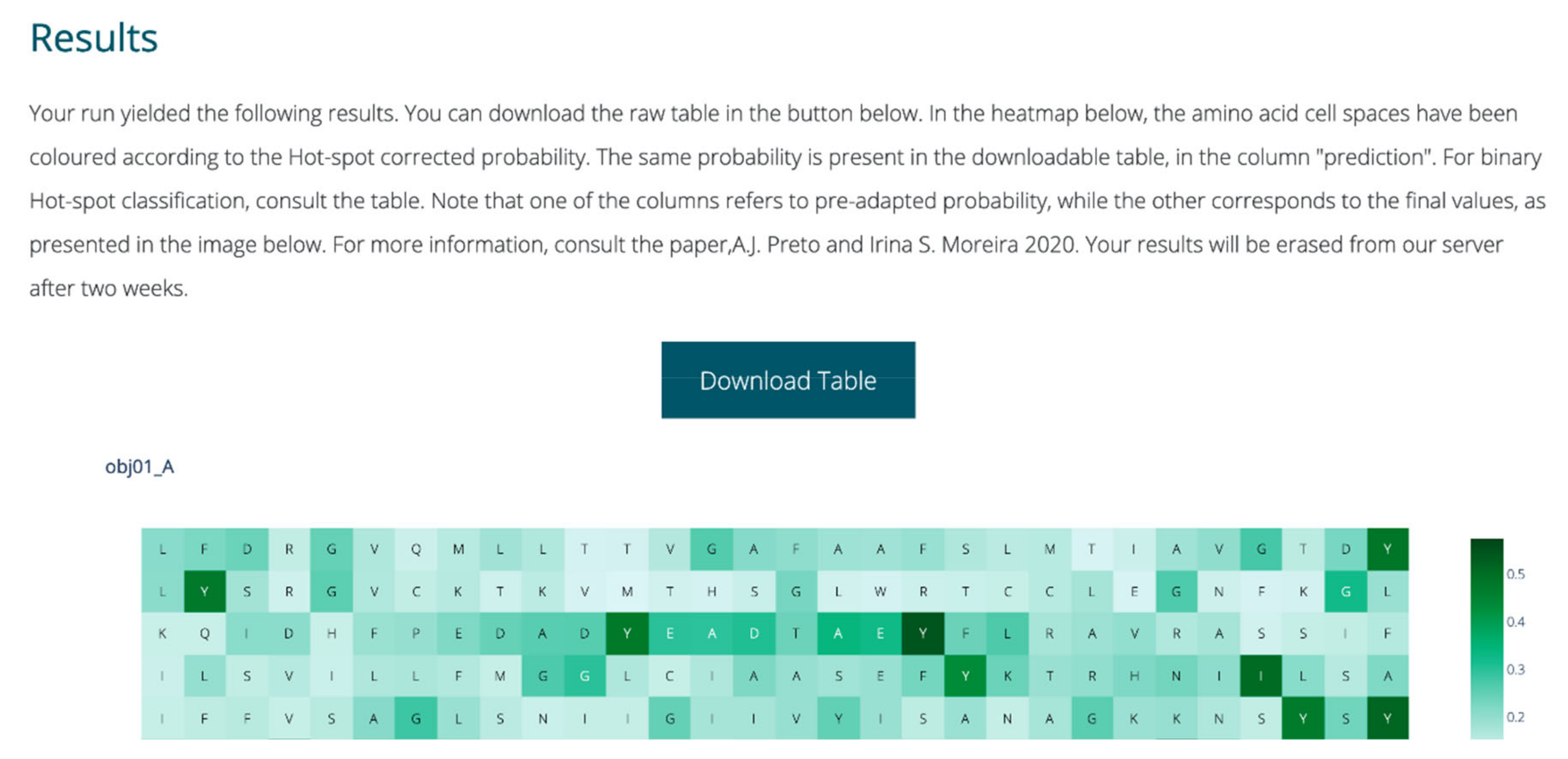
4.9. Webserver Implementation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data and Code Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
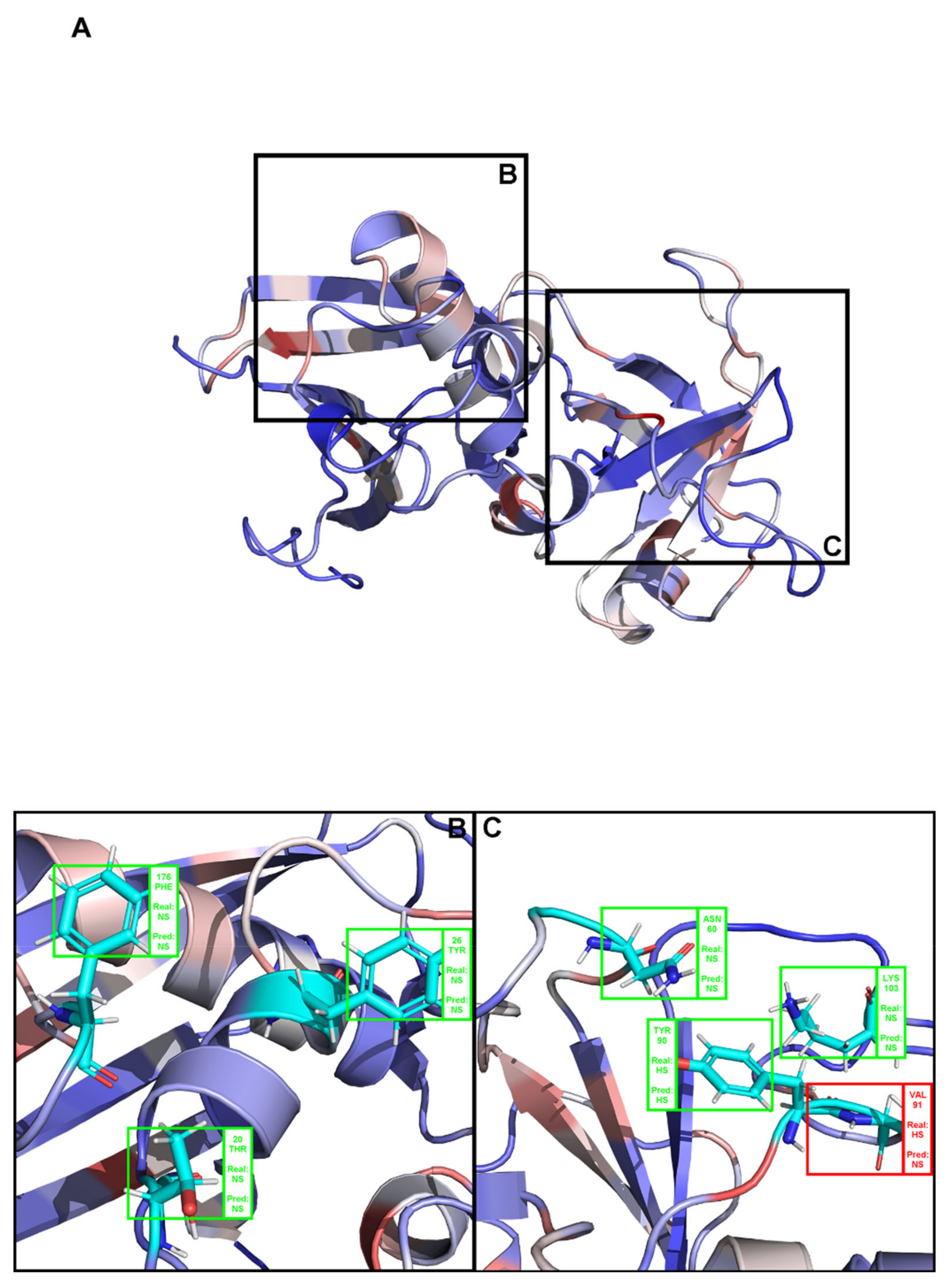{kind=link}
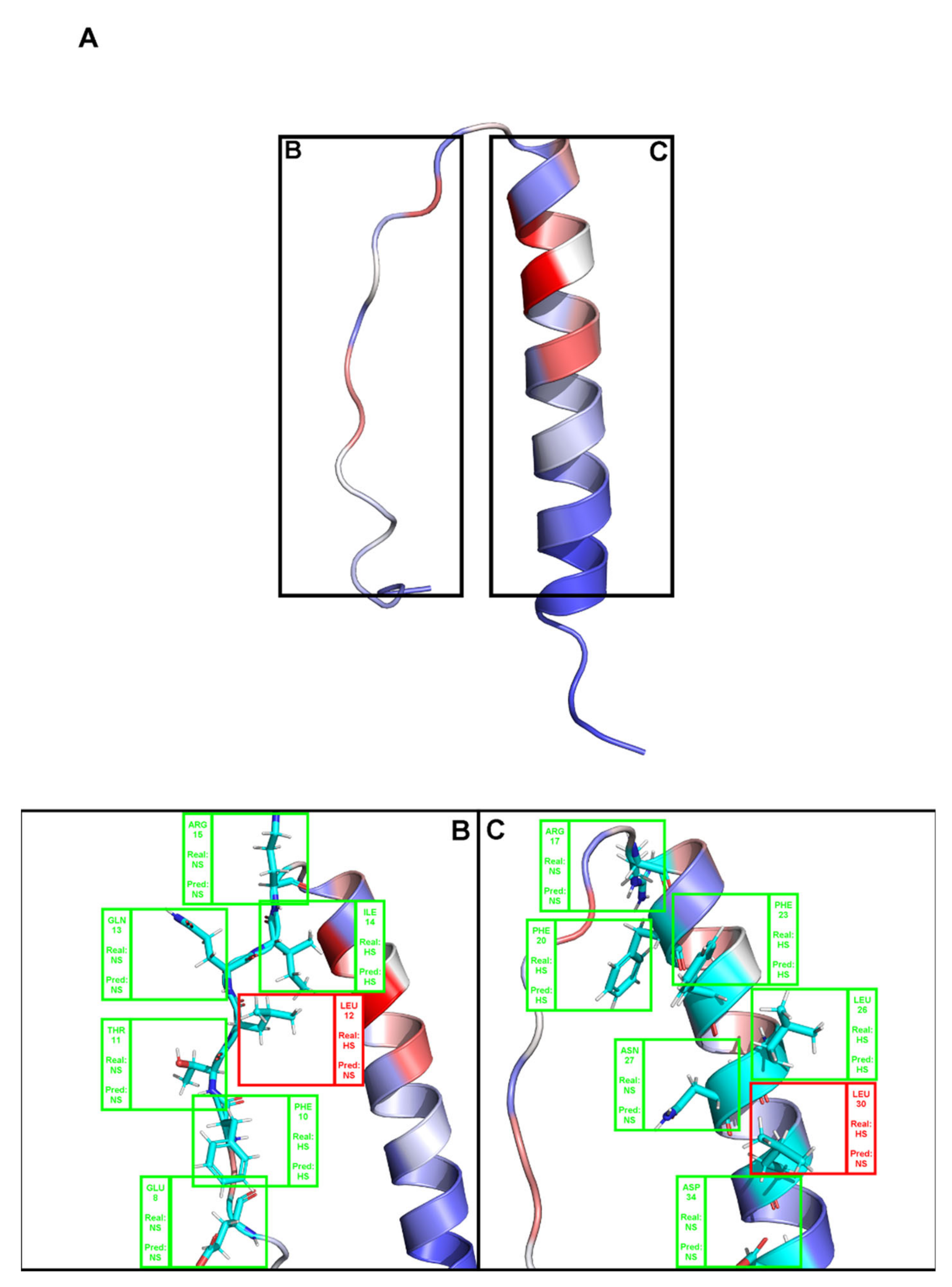{kind=link}
| Dataset | Classifier Name | Subset | Accuracy | AUC | Precision | Recall | F1-Score |
|---|---|---|---|---|---|---|---|
| In-house features | Extremely Randomized Trees | Train | 0.99 | 1.00 | 0.99 | 0.99 | 0.99 |
| Test | 0.81 | 0.77 | 0.88 | 0.81 | 0.83 | ||
| Neural Network | Train | 0.81 | 0.73 | 0.81 | 0.81 | 0.81 | |
| Test | 0.69 | 0.56 | 0.72 | 0.69 | 0.71 | ||
| AdaBoost | Train | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | |
| Test | 0.71 | 0.56 | 0.77 | 0.71 | 0.74 | ||
| Support Vector Machine | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | ||
| PSSM features | Extremely Randomized Trees | Train | 0.96 | 0.98 | 0.97 | 0.96 | 0.97 |
| Test | 0.72 | 0.55 | 0.82 | 0.72 | 0.76 | ||
| Neural Network | Train | 0.96 | 0.97 | 0.96 | 0.96 | 0.96 | |
| Test | 0.70 | 0.57 | 0.74 | 0.70 | 0.72 | ||
| AdaBoost | Train | 0.91 | 0.92 | 0.93 | 0.91 | 0.91 | |
| Test | 0.73 | 0.60 | 0.79 | 0.73 | 0.75 | ||
| Support Vector Machine | Train | 0.80 | 0.86 | 0.96 | 0.8 | 0.86 | |
| Test | 0.76 | 0.64 | 0.92 | 0.76 | 0.82 | ||
| In-house + PSSM | Extremely Randomized Trees | Train | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Test | 0.83 | 0.86 | 0.93 | 0.83 | 0.86 | ||
| Neural Network | Train | 0.83 | 0.78 | 0.85 | 0.83 | 0.82 | |
| Test | 0.56 | 0.50 | 0.52 | 0.56 | 0.53 | ||
| AdaBoost | Train | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 | |
| Test | 0.72 | 0.60 | 0.74 | 0.72 | 0.73 | ||
| Support Vector Machine | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 |
| Dataset | Classifier Name | Subset | Accuracy | AUC | Precision | Recall | F1-Score |
|---|---|---|---|---|---|---|---|
| In-house + iFeatures | Extremely Randomized Trees | Train | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Test | 0.83 | 0.77 | 0.85 | 0.83 | 0.84 | ||
| Neural Network | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | ||
| AdaBoost | Train | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | |
| Test | 0.81 | 0.75 | 0.83 | 0.81 | 0.82 | ||
| Support Vector Machine | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | ||
| In-house + PSSM + iFeatures | Extremely Randomized Trees | Train | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Test | 0.83 | 0.77 | 0.84 | 0.83 | 0.83 | ||
| Neural Network | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | ||
| AdaBoost | Train | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 | |
| Test | 0.77 | 0.69 | 0.79 | 0.77 | 0.78 | ||
| Support Vector Machine | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | ||
| iFeatures | Extremely Randomized Trees | Train | 0.83 | 0.80 | 0.90 | 0.83 | 0.85 |
| Test | 0.77 | 0.67 | 0.82 | 0.77 | 0.79 | ||
| Neural Network | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | ||
| AdaBoost | Train | 0.83 | 0.76 | 0.86 | 0.83 | 0.84 | |
| Test | 0.79 | 0.72 | 0.80 | 0.79 | 0.79 | ||
| Support Vector Machine | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 | |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 |
| Amino Acid | Adaptation Value | Accuracy | Precision | Recall | Amount Used for Threshold Adaptation | |
|---|---|---|---|---|---|---|
| ASP | Original | - | 0.71 | 0.00 | 1.00 | 11 |
| Adapted | - | - | - | |||
| SER | Original | - | 1.00 | 0.00 | 0.00 | 4 |
| Adapted | - | - | - | |||
| GLN | Original | - | 0.67 | 0.00 | 0.00 | 6 |
| Adapted | - | - | - | |||
| LYS | Original | - | 1.00 | 1.00 | 1.00 | 12 |
| Adapted | - | - | - | |||
| ILE | Original | +0.15 | 0.80 | 0.00 | 0.00 | 5 |
| Adapted | 1.00 | 1.00 | 1.00 | |||
| PRO | Original | +0.15 | 0.50 | 0.00 | 0.00 | 2 |
| Adapted | 1.00 | 1.00 | 1.00 | |||
| THR | Original | - | 1.00 | 1.00 | 1.00 | 8 |
| Adapted | - | - | - | |||
| PHE | Original | +0.25 | 0.75 | 0.00 | 0.00 | 4 |
| Adapted | 1.00 | 1.00 | 1.00 | |||
| ASN | Original | +0.15 | 0.50 | 0.00 | 0.00 | 6 |
| Adapted | 0.83 | 0.67 | 1.00 | |||
| GLY | Original | - | 1.00 | 0.00 | 0.00 | 1 |
| Adapted | - | - | - | |||
| HIS | Original | - | 0.80 | 0.00 | 0.00 | 5 |
| Adapted | - | - | - | |||
| LEU | Original | +0.06 | 0.50 | 0.00 | 0.00 | 4 |
| Adapted | 1.00 | 1.00 | 1.00 | |||
| ARG | Original | - | 1.00 | 0.00 | 0.00 | 9 |
| Adapted | - | - | - | |||
| TRP | Original | - | 0.71 | 0.00 | 0.00 | 7 |
| Adapted | - | - | - | |||
| VAL | Original | +0.25 | 0.67 | 0.00 | 0.00 | 3 |
| Adapted | 0.67 | 0.00 | 0.00 | |||
| GLU | Original | - | 0.85 | 0.00 | 0.00 | 13 |
| Adapted | - | - | - | |||
| TYR | Original | - | 0.55 | 0.33 | 0.67 | 11 |
| Adapted | - | - | - | |||
| Amino Acid | Helix Propensity | Sheet Propensity | Helix Propensity Values | Sheet Propensity Values | Molecular Weight | pKa Carboxylate | pKa Amine | pKa Side Chain | Number of Carbons |
|---|---|---|---|---|---|---|---|---|---|
| ALA | 1 | 1 | 1.45 | 0.97 | 89.09 | 2.30 | 9.90 | 0.00 | 3 |
| CYS | 2 | 2 | 0.77 | 1.30 | 121.16 | 1.80 | 10.80 | 8.65 | 3 |
| ASP | 2 | 3 | 0.98 | 0.80 | 133.10 | 2.00 | 10.00 | 4.04 | 4 |
| GLU | 1 | 4 | 1.53 | 0.26 | 147.13 | 2.20 | 9.70 | 4.39 | 5 |
| PHE | 3 | 2 | 1.12 | 1.28 | 165.19 | 1.80 | 9.10 | 0.00 | 9 |
| GLY | 4 | 3 | 0.53 | 0.81 | 75.07 | 2.40 | 9.80 | 0.00 | 2 |
| HIS | 3 | 5 | 1.24 | 0.71 | 155.16 | 1.80 | 9.20 | 6.75 | 6 |
| ILE | 5 | 6 | 1.00 | 1.60 | 131.17 | 2.40 | 9.70 | 0.00 | 6 |
| LYS | 5 | 5 | 1.07 | 0.74 | 146.19 | 2.20 | 9.20 | 11.00 | 6 |
| LEU | 1 | 2 | 1.34 | 1.22 | 131.17 | 2.40 | 9.60 | 0.00 | 6 |
| MET | 3 | 6 | 1.20 | 1.67 | 149.21 | 2.30 | 9.20 | 0.00 | 5 |
| ASN | 6 | 5 | 0.73 | 0.65 | 132.12 | 2.00 | 8.80 | 0.00 | 4 |
| PRO | 4 | 5 | 0.59 | 0.62 | 115.13 | 2.00 | 10.60 | 0.00 | 5 |
| GLN | 3 | 2 | 1.17 | 1.23 | 146.15 | 2.20 | 9.10 | 0.00 | 5 |
| ARG | 2 | 3 | 0.79 | 0.90 | 174.20 | 1.80 | 9.00 | 12.50 | 6 |
| SER | 2 | 5 | 0.79 | 0.72 | 105.09 | 2.10 | 9.20 | 0.00 | 3 |
| THR | 2 | 2 | 0.82 | 1.20 | 119.12 | 2.60 | 10.40 | 0.00 | 4 |
| VAL | 3 | 6 | 1.14 | 1.65 | 117.15 | 2.30 | 9.60 | 0.00 | 5 |
| TRP | 3 | 2 | 1.14 | 1.19 | 204.22 | 2.40 | 9.40 | 0.00 | 11 |
| TYR | 6 | 2 | 0.61 | 1.29 | 181.19 | 2.20 | 9.10 | 9.75 | 9 |
| Amino Acid | Number of Hydrogens | Number of Nitrogen Atoms | Number of Oxygens | Number of Sulphur | Standard Free Area | Protein Standard Area | Folded Buried Area | Mean Fractional Area | Residue Mass | Monoisotopic Mass |
|---|---|---|---|---|---|---|---|---|---|---|
| ALA | 7 | 1 | 2 | 0 | 118.10 | 31.50 | 86.60 | 0.74 | 71.08 | 71.04 |
| CYS | 7 | 1 | 2 | 1 | 146.10 | 13.90 | 132.30 | 0.91 | 103.14 | 103.01 |
| ASP | 7 | 1 | 4 | 0 | 158.70 | 60.90 | 97.80 | 0.62 | 115.09 | 115.03 |
| GLU | 9 | 1 | 4 | 0 | 186.20 | 72.30 | 113.90 | 0.62 | 129.12 | 129.04 |
| PHE | 11 | 1 | 2 | 0 | 222.80 | 28.70 | 194.10 | 0.88 | 147.18 | 147.07 |
| GLY | 5 | 1 | 2 | 0 | 88.10 | 25.20 | 62.90 | 0.72 | 57.05 | 57.02 |
| HIS | 9 | 3 | 2 | 0 | 202.50 | 46.70 | 155.80 | 0.78 | 137.14 | 137.06 |
| ILE | 13 | 1 | 2 | 0 | 181.00 | 23.00 | 158.00 | 0.88 | 113.16 | 113.08 |
| LYS | 14 | 2 | 2 | 0 | 225.80 | 110.30 | 115.50 | 0.52 | 128.17 | 128.10 |
| LEU | 13 | 1 | 2 | 0 | 193.10 | 29.00 | 164.10 | 0.85 | 113.16 | 113.08 |
| MET | 11 | 1 | 2 | 1 | 203.40 | 30.50 | 172.90 | 0.85 | 131.19 | 131.04 |
| ASN | 8 | 2 | 3 | 0 | 165.50 | 62.20 | 103.30 | 0.63 | 114.10 | 114.04 |
| PRO | 9 | 1 | 2 | 0 | 146.80 | 53.70 | 92.90 | 0.64 | 97.12 | 97.05 |
| GLN | 10 | 2 | 3 | 0 | 193.20 | 74.00 | 119.20 | 0.62 | 128.13 | 128.06 |
| ARG | 14 | 4 | 2 | 0 | 256.00 | 93.80 | 162.20 | 0.64 | 156.19 | 156.10 |
| SER | 7 | 1 | 3 | 0 | 129.80 | 44.20 | 85.60 | 0.66 | 87.08 | 87.03 |
| THR | 9 | 1 | 3 | 0 | 152.50 | 46.00 | 106.50 | 0.70 | 101.11 | 101.05 |
| VAL | 11 | 1 | 2 | 0 | 164.50 | 23.50 | 141.00 | 0.86 | 99.13 | 99.07 |
| TRP | 12 | 2 | 2 | 0 | 266.30 | 41.70 | 224.60 | 0.85 | 186.21 | 186.08 |
| TYR | 11 | 1 | 3 | 0 | 236.80 | 59.10 | 177.70 | 0.76 | 163.18 | 163.06 |
| Parameter | Default Value | Tested Values |
|---|---|---|
| n_estimators | 100 | (50,100,250,500,1000) |
| boostrap | False | (True, False) |
| class_weight | None | (None,”balanced_subsample”,”balanced”) |
| criterion | “gini” | (“gini”,”entropy”) |
| max_depth | None | (None,1,2,3) |
| min_samples_split | 2 | (2,3,4,5) |
| min_samples_leaf | 1 | (1,2,3) |
| max_leaf_nodes | None | (None,1,2,3) |
| max_samples | None | (None,1,2,5,10) |
| max_features | “auto” | (“auto”,”sqrt”,”log2”) |
| min_impurity_decrease | 0.0 | (0.0, 0.01, 0.001) |
| min_weight_fraction_leaf | 0.0 | (0.0, 0.01, 0.001) |



References
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Hot Spots—A Review of the Protein-Protein Interface Determinant Amino-Acid Residues. Proteins Struct. Funct. Genet. 2007, 68, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskin, O.; Ma, B.; Nussinov, R. Hot Regions in Protein-Protein Interactions: The Organization and Contribution of Structurally Conserved Hot Spot Residues. J. Mol. Biol. 2005, 345, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, N.; Chen, P.; Zheng, C.; Wang, B. Prediction of Protein Hotspots from Whole Protein Sequences by a Random Projection Ensemble System. Int. J. Mol. Sci. 2017, 18, 1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Xiong, Y.; Gao, H.; Zhu, X.; Chen, P. Protein-protein interface hot spots prediction based on hybrid feature selection strategy. BMC Bioinform. 2018, 19. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J.A. A hot spot of binding energy in a hormone-receptor interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef]
- Golden, M.S.; Cote, S.M.; Sayeg, M.; Zerbe, B.S.; Villar, E.A.; Beglov, D.; Sazinsky, S.L.; Georgiadis, R.M.; Vajda, S.; Kozakov, D.; et al. Comprehensive Experimental and Computational Analysis of Binding Energy Hot Spots at the NF-ΚB Essential Modulator/IKKβ Protein-Protein Interface. J. Am. Chem. Soc. 2013, 135, 6242–6256. [Google Scholar] [CrossRef] [Green Version]
- Ciglia, E.; Vergin, J.; Reimann, S.; Smits, S.H.J.; Schmitt, L.; Groth, G.; Gohlke, H. Resolving Hot Spots in the C-Terminal Dimerization Domain That Determine the Stability of the Molecular Chaperone Hsp90. PLoS ONE 2014, 9, e96031. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.H.; Tochowicz, A.; Pozzi, C.; Cardinale, D.; Ferrari, S.; Boum, Y.; Mangani, S.; Stroud, R.M.; Saxena, P.; Myllykallio, H.; et al. Hotspots in an Obligate Homodimeric Anticancer Target. Structural and Functional Effects of Interfacial Mutations in Human Thymidylate Synthase. J. Med. Chem. 2015, 58, 3572–3581. [Google Scholar] [CrossRef] [Green Version]
- Moreira, I.S. The Role of Water Occlusion for the Definition of a Protein Binding Hot-Spot. Curr. Top. Med. Chem. 2015, 15, 2068–2079. [Google Scholar] [CrossRef]
- Ramos, R.M.; Fernandes, L.F.; Moreira, I.S. Extending the applicability of the O-ring theory to protein-DNA complexes. Comput. Biol. Chem. 2013, 44, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Thorn, K.S.; Bogan, A.A. ASEdb: A Database of Alanine Mutations and Their Effects on the Free Energy of Binding in Protein Interactions. Bioinformatics 2001, 17, 284–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, T.B.; Arunachalam, K.V.; Bailey, D.; Mangual, V.; Bakhru, S.; Russo, R.; Huang, D.; Paczkowski, M.; Lalchandani, V.; Ramachandra, C.; et al. The Binding Interface Database (BID): A Compilation of Amino Acid Hot Spots in Protein Interfaces. Bioinformatics 2003, 19, 1453–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.D.S.; Gromiha, M.M. PINT: Protein-Protein Interactions Thermodynamic Database. Nucleic Acids Res. 2006, 34, D195–D198. [Google Scholar] [CrossRef]
- Moal, I.H.; Fernandez-Recio, J. SKEMPI: A Structural Kinetic and Energetic Database of Mutant Protein Interactions and Its Use in Empirical Models. Bioinformatics 2012, 28, 2600–2607. [Google Scholar] [CrossRef] [Green Version]
- Jankauskaite, J.; Jiménez-García, B.; Dapkunas, J.; Fernandéz-Recio, J.; Moal, I.H. SKEMPI 2.0: And updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 2019, 35, 462–469. [Google Scholar] [CrossRef]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot Spots in Protein–Protein Interfaces: Towards Drug Discovery. Prog. Biophys. Mol. Biol. 2014, 116, 165–173. [Google Scholar] [CrossRef]
- Morrow, J.K.; Zhang, S. Computational Prediction of Protein Hot Spot Residues. Curr. Pharm. Des. 2012, 18, 1255–1265. [Google Scholar] [CrossRef]
- Nguyen, Q.; Fablet, R.; Pastor, D. Protein Interaction Hotspot Identification Using Sequence-Based Frequency-Derived Features. IEEE Trans. Biomed. Eng. 2013, 60, 2993–3002. [Google Scholar] [CrossRef]
- Hu, S.-S.; Chen, P.; Wang, B.; Li, J. Protein Binding Hot Spots Prediction from Sequence Only by a New Ensemble Learning Method. Amino Acids 2017, 49, 1773–1785. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, P.; Wang, B.; Zhang, J.; Li, J. Hot Spot Prediction in Protein-Protein Interactions by an Ensemble System. BMC Syst. Biol. 2018, 12 (Suppl. 9), 132. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Koukos, P.I.; Melo, R.; Almeida, J.G.; Preto, A.J.; Schaarschmidt, J.; Trellet, M.; Gümüş, Z.H.; Costa, J.; Bonvin, A.M.J.J. SpotOn: High Accuracy Identification of Protein-Protein Interface Hot-Spots. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.M.; Ramos, R.M.; Pimenta, A.C.; Moreira, I.S. Solvent-Accessible Surface Area: How Well Can Be Applied to Hot-Spot Detection? Proteins Struct. Funct. Bioinforma. 2014, 82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Mitchell, J.C. KFC2: A Knowledge-Based Hot Spot Prediction Method Based on Interface Solvation, Atomic Density, and Plasticity Features. Proteins 2011, 79, 2671–2683. [Google Scholar] [CrossRef]
- Tuncbag, N.; Keskin, O.; Gursoy, A. HotPoint: Hot Spot Prediction Server for Protein Interfaces. Nucleic Acids Res. 2010, 38, W402–W406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Cryst. Sect. D Biol. Cryst. 2002, 28, 235–242. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.-C.; et al. IFeature: A Python Package and Web Server for Features Extraction and Selection from Protein and Peptide Sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36 (Suppl. 1), D402–D408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, G.E. Connectionist Learning Procedures. Artif. Intell. 1989, 40, 185–234. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.-F.; Lin, C.-J.; Weng, R.C. Probability Estimates for Multi-Class Classification by Pairwise Coupling. J. Mach. Learn. Res. 2004, 5, 975–1005. [Google Scholar]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely Randomized Trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. iGHBP: Computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput. Struct. Biotechnol. J. 2018, 16, 412–420. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Wei, L.; Lee, G. AtbPpred: A Robust Sequence-Based Prediction of Anti-Tubercular Peptides Using Extremely Randomized Trees. Comput. Struct. Biotechnol. J. 2019, 17, 972–981. [Google Scholar] [CrossRef]
- Plotly Technologies Inc. Collaborative Data Science; Plotly Technologies Inc.: Montreal, QC, Canada, 2015. [Google Scholar]
- Grinberg, M. Flask Web Development: Developing Web Applications with Python, 1st ed.; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2014. [Google Scholar]



| Method | Data | Acc | AUROC | Prec | Rec | F1 |
|---|---|---|---|---|---|---|
| Neural network | Train | 0.81 | 0.73 | 0.81 | 0.81 | 0.81 |
| Test | 0.69 | 0.56 | 0.72 | 0.69 | 0.71 | |
| AdaBoost | Train | 0.98 | 0.98 | 0.98 | 0.98 | 0.98 |
| Test | 0.71 | 0.56 | 0.77 | 0.71 | 0.74 | |
| Support Vector Machine | Train | 0.77 | 0.00 | 1.00 | 0.77 | 0.87 |
| Test | 0.76 | 0.00 | 1.00 | 0.76 | 0.86 | |
| Extremely Randomized Trees | Train | 0.99 | 1.00 | 0.99 | 0.99 | 0.99 |
| Test | 0.81 | 0.77 | 0.88 | 0.81 | 0.83 |
| Data | Acc | AUROC | Prec | Rec | F1 |
|---|---|---|---|---|---|
| Training after threshold adaptation | 0.99 | 0.99 | 0.99 | 0.99 | 0.99 |
| Testing after threshold adaptation | 0.85 | 0.88 | 0.93 | 0.85 | 0.87 |
| Independent Testing after threshold adaptation | 0.82 | 0.83 | 0.91 | 0.82 | 0.85 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preto, A.J.; Moreira, I.S. SPOTONE: Hot Spots on Protein Complexes with Extremely Randomized Trees via Sequence-Only Features. Int. J. Mol. Sci. 2020, 21, 7281. https://doi.org/10.3390/ijms21197281
Preto AJ, Moreira IS. SPOTONE: Hot Spots on Protein Complexes with Extremely Randomized Trees via Sequence-Only Features. International Journal of Molecular Sciences. 2020; 21(19):7281. https://doi.org/10.3390/ijms21197281
Chicago/Turabian StylePreto, A. J., and Irina S. Moreira. 2020. "SPOTONE: Hot Spots on Protein Complexes with Extremely Randomized Trees via Sequence-Only Features" International Journal of Molecular Sciences 21, no. 19: 7281. https://doi.org/10.3390/ijms21197281
APA StylePreto, A. J., & Moreira, I. S. (2020). SPOTONE: Hot Spots on Protein Complexes with Extremely Randomized Trees via Sequence-Only Features. International Journal of Molecular Sciences, 21(19), 7281. https://doi.org/10.3390/ijms21197281